

{kind=link}
{kind=link}
{kind=link}
{kind=link}
基于全基因组重测序信息开发玉米H99自交系特异分子标记
引用本文
吕远大, 李坦, 石丽, 张晓林, 赵涵. 基于全基因组重测序信息开发玉米H99自交系特异分子标记. 作物学报, 2014, 40(2): 191-197[L#cod#x000dc; Yuan-Da, LI Tan, SHI Li, ZHANG Xiao-Lin, ZHAO Han. Next-generation Sequencing for Molecular Marker Development in Maize Inbred H99. 作物学报, 2014, 40(2): 191-197] 
Permissions
Copyright©2014, 作物学报编辑部
作物学报编辑部
基于全基因组重测序信息开发玉米H99自交系特异分子标记
* 通讯作者(Corresponding author): 赵涵, E-mail:zhaohancn@gmail.com
**同等贡献(Contributed equally to this work)
摘要
随着基因组测序技术和计算生物学的发展, 利用生物信息学的方法大规模挖掘基因组特异分子标记已成为可能。玉米自交系H99具有较强的可再生能力, 是玉米转基因研究的主要供体材料, 但是目前对H99基因组信息了解甚少。本研究利用高通量Illumina测序技术, 对H99全基因组进行重测序, 利用生物信息学手段大规模挖掘并开发了4043个H99特异性的SSR分子标记。随后利用模拟PCR策略, 对开发的SSR分子标记进行电子多态性筛选, 针对B73×H99、Mo17×H99和B73×Mo17三个群体亲本组合共开发2699候选的特异多态性SSR分子标记。随机挑选20对SSR分子标记对群体亲本B73、H99和Mo17进行多态性筛选, 进一步证实候选的多态性具有95%的准确率。此外, 基于B73参考序列对开发的多态性SSR分子标记进行全基因组染色体定位及基因注释, 揭示了候选多态性SSR分子标记在全基因组及基因内的分布特征。为玉米自交系H99开发了大量的分子标记资源, 同时也为快速开发品种间多态性分子标记提供了一套高效可行的分析方法。
关键词:
玉米H99自交系; 高通量测序; 电子PCR; 多态性SSR
Next-generation Sequencing for Molecular Marker Development in Maize Inbred H99
Abstract
Next Generation Sequencing (NGS) has provided an effective approach to reveal the large scale of DNA polymorphic loci used as molecular markers to distinguish the genetic variations among different genotypes. Maize inbred line H99 is a common transgenic genotype with its desirable regeneration capacity. However, the genome sequence of H99 is unsequenced, which lags the molecular marker development for further maker assisted selection. Here, we used next generation sequencing to resequence H99 whole genome. The contigs assembled with SOAPdenovo2 were further scanned for potential SSR loci by MISA software. Out of 8268 SSR loci, 4043 site-specific primers flanking SSR loci were designed and surveyed
Keyword:
Maize inbred line H99; High throughout sequencing; ePCR; Polymorphic SSR
分子标记是重要农艺性状基因的克隆和分子育种必不可少的工具。目前常用的分子标记主要有RFLP、RAPD、AFLP、SSR、STS和SNP等[ 1, 2, 3, 4]。其中, SSR标记具备多态性丰富、重复性好、易于使用、可检测位点多、共显性且均匀分布整个基因组等优点, 已成为遗传和育种研究中最为广泛应用的分子标记[ 3, 5]。利用玉米B73及Mo17杂交的F1群体内个体进行4代互交, 随后连续自交发展的重组自交系IBM群体, 用该群体已成功将上千个根据B73基因组中重复序列设计的SSR分子标记定位在玉米10条染色体上[ 6]。SSR分子标记已被广泛应用于其他玉米亲本组合连锁群构建、多样性分析、杂种纯度检测等研究中, 但是由于它们主要来源于B73基因组并应用于B73和Mo17的分离群体, 导致其多态性在其这些亲本组合中相对较低[ 7, 8, 9]。
近年来, 基因组测序技术的迅猛发展, 尤其是以Illumina公司发展的边合成边测序(sequencing by synthesis)为技术核心的第二代测序已被广泛应用于动植物全基因组测序、基因组重测序、转录组、sRNA和表观基因组等方面的研究[ 10, 11, 12, 13, 14, 15]。Lai实验室利用该技术对中国6个玉米优良自交系重测序后, 又对近250个自交系进行了重测序工作[ 16]。冷泉港实验室也对近200个从全世界搜集的玉米自交系测序, 将原始测序结果提交NCBI数据库[ 17]。通过生物信息学分析比较发现, 玉米不同基因型中存在着大量的序列变异, 其中SNP和小InDel的数量尤为突出。Lai等[ 16]估算中国6个玉米优良自交系的非重复区大约存在1 272 134个SNP和30 178个InDel, 其中约有 68 966个SNP和571个InDel位于功能基因内。这些序列的变异直接导致了玉米表现型的多样性, 同样也为揭示玉米重要农艺性状的遗传基础提供了标记位点。SNP标记被认为是高通量、低成本的分子标记, 但是由于芯片设计、试剂、检测仪器成本高等原因, 限制了其应用范围[ 4]。SSR与InDel则主要表现在序列长度的多态性。但是由于生物信息算法的局限性和测序深度较低等原因, 发现的InDel往往分布在1~10 bp范围内, 为多态性检测带来了一定的难度[ 18, 19, 20]。玉米中由于存在较多转座子, SSR位点的多态性表现得极为丰富且长度差异加大[ 21]。通过特异性引物的PCR扩增、琼脂糖凝胶电泳或聚丙烯酰胺凝胶电泳、放射自显影或银染技术等, 就可以检测到在简单重复序列重复次数不同的DNA区域的多态性。
与传统SSR分子标记的开发相比, 基于NGS高通量测序数据的大规模SSR开发, 因其高通量、高覆盖度、高密度及可操作性等优点而被应用于多个物种的分子标记资源开发。Zalapa等[ 22]提出了利用NGS高通量454测序技术开发植物SSR分子标记的策略, 利用大果越桔( Vaccinium macrocarpon) 454高通量测序数据开发了SSR分子标记, 随后通过实验检测了该方法的可行性和准确性。Yu等[ 23]利用454高通量测序数据快速高效开发了河鹿( Hydropotes inermis argyropus)的SSR分子标记。Wang等[ 24]利用Illumina高通量测序技术对菊花脑( Chrysanthemum nankingense)转录组进行Paired-end双末端测序, 并进一步开发了SSR分子标记。然而, 在高通量SSR分子标记开发的同时也带来了引物合成等实验成本的增加, 加上SSR分子标记的多态率普遍较低, 更造成了资源上的浪费和时间的损耗。Yonemaru等[ 25]利用高粱( Sorghum bicolor)全基因组鸟枪法测序数据开发了SSR分子标记, 进一步利用ePCR策略进行电子多态性预筛选, 提高了SSR标记的多态率和可用性。Lü等[ 26, 27]基于海岛棉( Gossypium barbadense)基因组数据资源, 先后利用ePCR策略对挖掘的SSR、InDel分子标记进行了多态性预筛选, 极大地增加了多态性分子标记资源, 同时也大大降低了实验成本。
本研究利用Illumina高通量基因组重测序技术, 及生物信息学手段, 准确、快速地开发了一套基于玉米H99基因型的SSR分子标记, 随后通过计算机模拟筛选出可用于B73×H99、Mo17×H99和B73× Mo173个群体的候选多态性SSR分子标记, 为其相关分子育种研究提供重要的基础, 同时也为发展分子标记提供一套高效的分析方法。
1 材料与方法
1.1 供试材料及全基因组测序
供试材料为B73、Mo 17、H99三种玉米自交系。用其叶片按照Karroten植物基因组DNA提取试剂盒相关步骤提取DNA。用0.8%的琼脂糖凝胶检测提取的DNA样本质量。H99全基因组重测序采用Illumina HiSeq 2000测序仪, 由上海翰宇生物技术有限公司完成。
1.2 序列清理及全基因组装
H99全基因组Illumina下机测序数据, 通过CASAVA 1.8.2 (
随后, 利用SOAPdenovo2[ 29]组装软件对清理后的序列进行后续的全基因组从头 de novo组装。SOAPdenovo2采用De Bruijn Graph算法, 组装对 K-mer值设置有较大的依赖。本研究中, 通过31~49共10个梯度值( K值须为奇数)分别组装, 最终根据组装后Scaffolds N50大小来选取最优 K-mer, 进而确定最优组装结果, 其余参数均为默认。
1.3 全基因组SSR挖掘及标记开发
利用MISA (http://pgrc.ipk-gatersleben.de/misa/) SSR挖掘程序搜索H99全基因组SSR信息, 进一步利用SSR侧翼序列设计PCR特异引物, 开发SSR分子标记。本研究中, SSR的查找标准为单核苷酸重复≥20, 二核苷酸重复次数≥10, 三核苷酸重复次数≥7, 四核苷酸重复次数≥5, 五核苷酸重复次数≥4, 六核苷酸重复次数≥4, 复合型的SSR整体长度不小于24 bp。SSR引物设计由Primer3程序完成[ 30]。引物设计的主要参数为引物长度18~20 bp, 最适 20 bp; PCR产物长度100~250 bp; Tm值45~55℃, 最适50℃; GC含量为45%~65%, 最适50%。所有引物均由上海生工生物科技有限公司合成。
1.4 多态性SSR分子标记电子筛选
鉴于B73、Mo17基因组序列资源的存在, 使得利用模拟PCR策略对H99 SSR标记引物进行电子多态性筛选成为可能。用所有SSR引物分别与B73、Mo17及H99基因组进行了模拟PCR扩增, 进一步通过ePCR软件(http://www.ncbi.nlm.nih.gov/ projects/e-pcr/)分析了SSR标记位点的多态性。利用本地数据库预测PCR在不同基因型材料之间的扩增长度, 模拟中碱基错配值Mismatch≤3 bp。随后, 通过电子多态性筛选的候选多态性SSR分子标记, 用于进一步的染色体定位和基因组注释。
1.5 全基因组定位及注释
为了进一步揭示SSR相关的基因组信息及变异效应, 利用所开发的SSR分子标记保守侧翼序列, 结合玉米B73 (V2)基因组序列及基因注释信息, 采用SNPeff[ 31]全基因组注释程序对候选多态性标记进行物理定位和基因组注释。
1.6 PCR扩增和电泳多态性检测
为了进一步考察候选多态性SSR的准确性, 我们在每条染色体臂上共随机挑选20对新开发的SSR引物, 在B73、Mo17和H99基因组中进行了多态性验证。PCR反应总体系为25 μL, 包括10×buffer (含Mg2+) 2.5 μL, dNTP 2.5 μL, TaqDNA聚合酶0.1 μL, 引物2 μL, 模板DNA 2 μL, ddH2O 15.9 μL, 上覆 20 μL矿物油。PCR反应程序为, 94℃预变性3 min, 94℃变性30 s, 58℃退火1 min, 72℃延伸80 s, 共进行38个循环。用2%琼脂糖凝胶加压100 V电泳, 紫外透射仪上观察、照相。
2 结果与分析
2.1 H99全基因组测序及 de novo组装
利用Illumina Paired-End双端100 bp×2测序, 共获得21 Gb玉米H99基因组数据。NCBI中SRA数据库释放了一组Paired-End 150 bp×2的H99基因组数据(SRR671686), 总碱基数约1.9 Gb, 合计约23 Gb碱基量, 约覆盖10×基因组。随后, 通过Q20质量过滤和L20长度清理, 数据碱基数约22 Gb, 平均序列长度98.4 bp (表1)。证实了H99基因组数据具有较高的测序质量。
| 表1 H99全基因组测序数据汇总 Table 1 Summary of H99 whole-genome sequencing |
清理后数据经SOAPdenovo2组装程序, de novo从头拼接, 通过多k-mer值(31-49)组装筛选, 根据N50大小筛选出最优k-mer为43, 确定为最优组装结果。组装后共获得2 894 673条Contig (≥100 bp), N50为200 bp, 最长的Contig为6594 bp, 总共拼接的长度为570 456 877 bp。利用Paired-end信息, 进一步构建Scaffolds后, 获得186 042条Scaffolds序列, 最长Scaffold为8258 bp, N50为236 bp, 共 128 707 495 bp (表1)。随后, ≥400 bp的Scaffolds序列被用于后续的SSR挖掘。
2.2 SSR发掘及分子标记开发
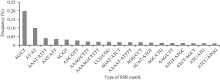
利用MISA软件搜寻含1~6个重复基元的SSR区间, 共获得8268个SSR位点, 分布于8139条Contigs序列中。其中, 单一重复类型(单个重复基元) SSR有8200个(99.18%), 而复合型(多个重复基元) SSR则有68个(0.82%)。重复基元分析显示, 分布最多的重复基元为二核苷酸重复, 占32.99%, 随后五核苷酸重复占22.08%, 四核苷酸重复占20.26%, 三核苷酸重复占15.04%, 六核苷酸重复占9.15%以及单核苷酸重复仅0.47%。此外, 重复基元类型分析结果显示, 8268个SSR中共有368种重复基元(Motif), 其中, 所占比例最大的是AG/CT (19.77%), 其次为AT占9.98%, AAAT/ATTT占4.06% (图1)。
进一步利用Primer3程序, 针对SSR位点两端侧翼序列, 共设计出4043对SSR引物, 用于后续分析。
 | 图1 SSR重复基元类型分布频率Fig. 1 Frequency of SSR motif types |
2.3 电子多态性筛选
鉴于B73、Mo17基因组序列资源的存在, 使得利用ePCR策略对H99 SSR标记引物进行电子多态性筛选成为可能。4043对SSR引物分别与B73、Mo17及H99基因组序列进行了ePCR扩增, 进一步分析了标记位点的多态性。结果发现, B73×H99间差异的有2223个候选位点, ≥10 bp的标记有1023个; Mo17×H99间有1684个候选多态性位点, ≥10 bp有746个; B73×Mo17间则有1427个多态性位点, ≥10 bp的有664个(图2)。因此, 针对不同群体组合及电子多态性统计结果, 选取多态片段≥10 bp, 分别针对B73×H99、Mo17×H99和B73×Mo17群体开发了3套候选的多态性SSR标记。此外, 826个SSR分子标记同时适用于3个群体中, 呈现了较好的通用性。
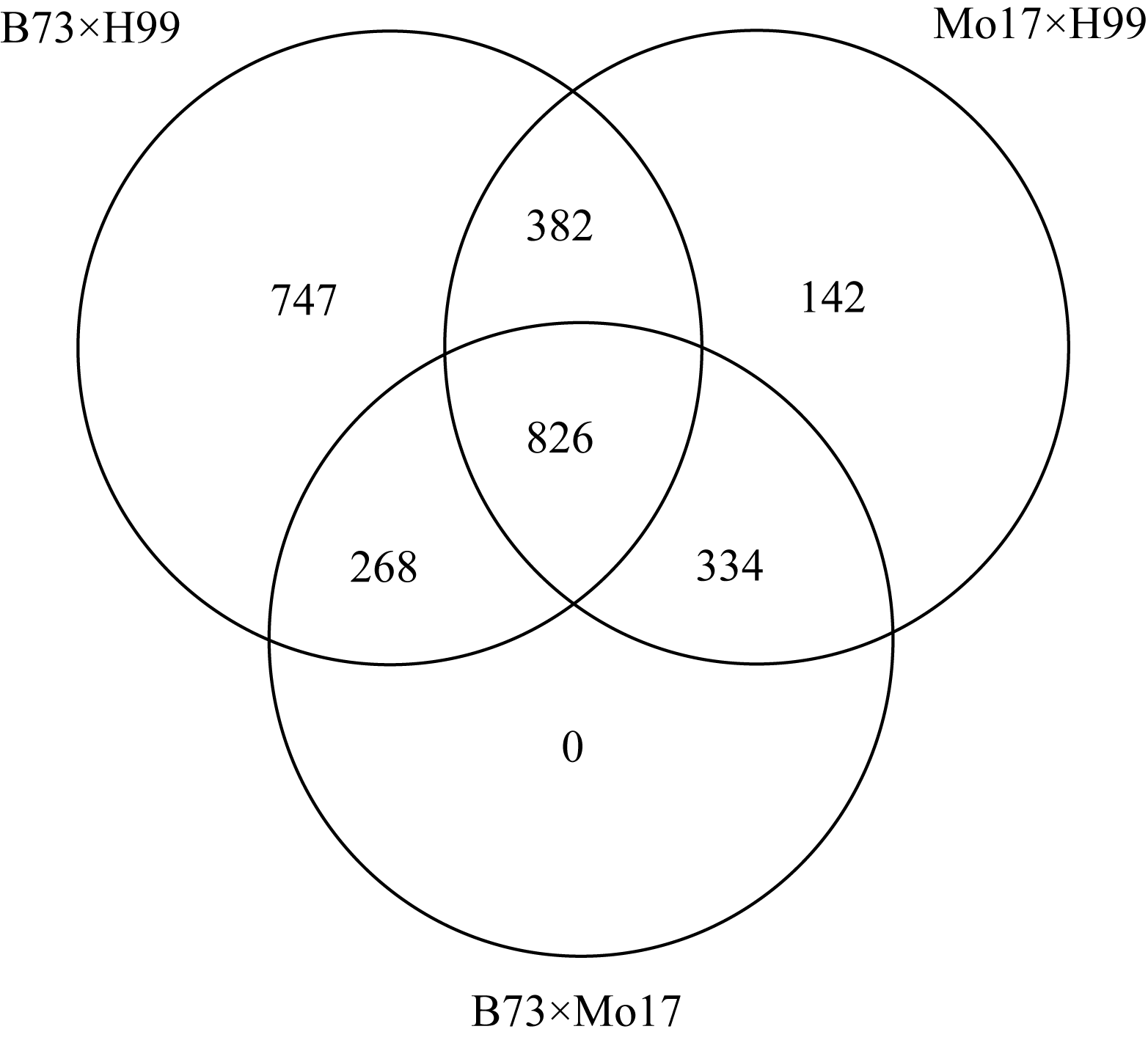 | 图2 分别用于B73×H99、Mo17×H99和B73×Mo17群体的候选多态性SSR标记Fig. 2 Candidate polymorphic SSR markers for B73×H99, Mo17×H99, and B73×Mo17 populations |
详细的候选多态性分子标记引物信息可见网址(ftp://ftp.maizegenes.org/pub/H99paper_S1.xls)。
2.4 候选多态性SSR基因组定位及注释
基于B73参考基因组, 利用SNPeff V3基因组注释程序, 对3套候选的多态性SSR合计2699分子标记进行了染色体定位和基因组注释。染色体定位结果显示, 在2699个新开发的SSR标记中, 2557个SSR标记被成功定位到B73参考基因组上, 其中2074 (76.84%) 特异性地定位到一个位点, 仅有483 (17.90%)定位到多个位点, 而余下142 (0.53%)则未能定位到玉米B73 (V2)参考基因组。此外, 有18个SSR被定位在未知区域, 缺少染色体信息, 1个SSR被定位在线粒体中。除预测的着丝粒位置附近, SSR引物位置在染色体上基本呈均匀分布。详细的SSR标记物理定位图见网址(ftp://ftp.maizegenes.org/ pub/H99paper_S2. pdf)。
进一步通过全基因组注释, 2074 SSR标记特异性定位位点中, 非编码区3° UTR存在92个, 5° UTR存在101个; 内含子区存在360个; 外显子区存在186个; 调控区(启动子区Promoter-TSS有427个; TTS有303个); 基因间区含605个(图3)。而483 SSR标记定位多达6052个位点, 这些SSR标记往往涉及多个基因信息。详细的基因组定位及注释信息见网址(ftp://ftp.maizegenes.org/pub/H99paper_S3.xls)。
2.5 SSR分子标记多态性检测
为了进一步考察候选多态性SSR的准确性, 在每条染色体臂上随机挑选一个新开发引物, 共计20对, 在B73、Mo17和H99基因组中进行了多态性验证。通过琼脂糖凝胶电泳发现, 在检测的20对SSR引物中, 19对引物可以清楚揭示B73、Mo17和H99基因组DNA之间的多态, 多态率达90% (图4)。同时, PCR产物片段大小与ePCR预测结果完全吻合。这一结果进一步证实了通过ePCR模拟策略预测分子标记多态性的准确性和可行性。
 | 图3 基于B73基因组的SSR基因组注释分析Fig. 3 Variant annotation of SSR based on B73 gene set |
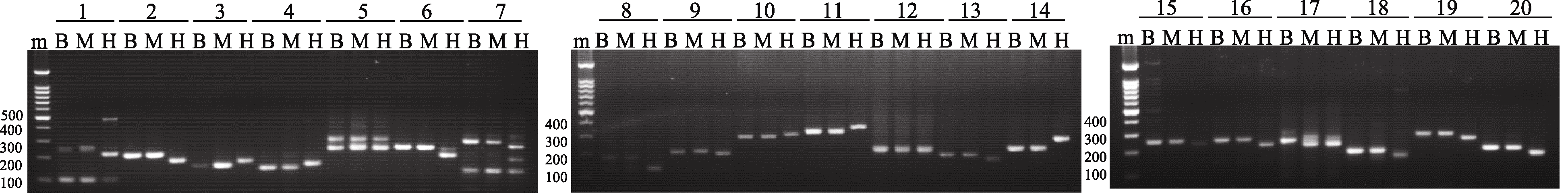 | 图4 3个玉米自交系(B73、Mo17和H99)中20对SSR引物基因组扩增的电泳图Fig. 4 Banding patterns of 20 SSR primer pairs in three maize inbred lines (B73, Mo17, and H99) m: marker; B: B73; M: Mo17; H:H99. |
3 讨论
玉米自交系H99具有较好的二型愈伤诱导率, 已被广泛作为农杆菌介导转基因受体亲本[ 32]。但是H99配合力差, 很难直接作为杂交种亲本, 只有使用其他自交系作为轮回亲本, 结合前景和背景选择, 才能把目的基因快速转入优良自交系中[ 33]。背景选择的关键是要获得足够的均匀分布在全基因组的分子标记。本研究利用Illumina HiSeq 2000对H99自交系10倍理论覆盖率进行全基因组测序, 经过一系列的优化组装, SSR位点扫描, 分别针对3个群体B73×H99、Mo17×H99以及B73×Mo17亲本基因组序列进行SSR电子多态性筛选, 共开发了2699个分布于玉米10条染色体的候选的多态性SSR标记引物。
高通量测序技术的发展以及数据分析软件的不断完善, 利用二代测序结果发展各种分子标记已经成为可能[ 11, 15]。NCBI目前已公布近400个玉米自交系重测序结果, 通过简单的过滤、比对、拼接及位点查找, 可以很快找到自交系组合间多态性位点, 如SNP、InDel、SSR等。据估计, 玉米每76个碱基就存在一个SNP位点, 但由于受到检测费用的限制, SNP分子标记主要应用于遗传图谱构建, 全基因组关联分析(GWAS)等群体单株数目相对较少的遗传研究, 而很少用于遗传育种中大群体样本选择过程, 如分子标记辅助遗传育种中的背景选择[ 34, 35, 36, 37, 38]。InDel在玉米基因组中也广泛分布, 但是受重测序深度, 算法等因素限制, 目前往往仅检测到小于10 bp的小InDel, 且假阳性率较高[ 39]。而且根据单个小InDel设计的分子标记检测起来较为困难。SSR序列多态性的出现是由于同一物种中不同基因型的寡聚核苷酸重复次数不同造成的, 而重复次数的多态性与染色体复制时的滑动或染色单体的不等交换相关。尽管SSR位点在玉米基因组上分布远不如SNP和InDel密集, 但是侧翼序列通常都是保守性较强的单一序列, 便于设计PCR引物。本研究利用SSR侧翼序列设计的引物, 在3种基因型DNA中扩增成功率达到了100%。
通过功能注释发现, 大多数候选的多态性SSR位点主要位于玉米功能基因区域(71%), 而分布在基因间区的标记约占29%。分布于功能基因区间的SSR分子标记由于与功能基因紧密相连, 可被开发成功能性分子标记, 显著提高分子标记对目的功能基因的选择效率。对玉米H99 SSR位点详细分析后发现, 仍然有SSR位点无法物理定位在玉米测序品种B73参考基因组上。究其原因, 一方面玉米B73基因组中仍存在大量的未测序区间, 另一方面玉米不同品种基因组中广泛存在着PAV (presence- absence variation)区段[ 16], 这些未能定位的SSR可能来自H99的特异性区域。
4 结论
利用玉米自交系H99 Illumina高通量重测序数据, 针对B73×H99、Mo17×H99和B73×Mo17三个群体亲本组合开发了各自特异多态性SSR分子标记, 为研究不同群体材料间遗传差异分析、分子遗传图谱构建、深入揭示基因的染色体分布特征及基因组变异等奠定了重要的基础。同时, 高通量测序技术结合多基因组交互ePCR的生物信息学分析策略, 为大规模开发基因型专化多态性SSR分子标记提供了重要的思路和研究方法。
The authors have declared that no competing interests exist.
作者已声明无竞争性利益关系。The authors have declared that no competing interests exist.
参考文献
| 1 |
|
| 2 |
|
| 3 |
|
| 4 |
|
| 5 |
|
| 6 |
|
| 7 |
|
| 8 |
|
| 9 |
|
| 10 |
|
| 11 |
|
| 12 |
|
| 13 |
|
| 14 |
|
| 15 |
|
| 16 |
|
| 17 |
|
| 18 |
|
| 19 |
|
| 20 |
|
| 21 |
|
| 22 |
|
| 23 |
|
| 24 |
|
| 25 |
|
| 26 |
|
| 27 |
|
| 28 |
|
| 29 |
|
| 30 |
|
| 31 |
|
| 32 |
|
| 33 |
|
| 34 |
|
| 35 |
|
| 36 |
|
| 37 |
|
| 38 |
|
| 39 |
|