{kind=link}
{kind=link}
{kind=link}
玉米行粒数的全基因组关联分析
[吴律 , 代力强, 董青松, 施婷婷, 王丕武
, 代力强, 董青松, 施婷婷, 王丕武* ]
, 代力强, 董青松, 施婷婷, 王丕武]
|
|


第一作者联系方式: E-mail: 13224341842@163.com
行粒数是玉米重要的产量构成性状之一, 对其遗传机理进行深入研究具有重要的理论和现实意义。本研究以吉林省80份核心玉米自交系作为关联群体, 于2014年和2015年分别在吉林省长春和梅河口进行行粒数测定。同时利用第2代测序技术对关联群体进行全基因组重测序, 获得的SNP标记用于后续分析。结果显示, 不同环境下玉米行粒数表型性状变异范围在12.0~41.6之间, 遗传力为36.4%。关联分析结果共得到19个与玉米行粒数显著关联的SNP标记, 其中位于染色体框2.04和3.08的两个标记在2015年长春和梅河口均被检测到, 14个SNP标记位于前人已定位到的QTL置信区间内。在显著性SNP标记的连锁不平衡区域内挖掘出4个候选基因, 分别预测编码泛素化目标受体蛋白、金属依赖性磷酸水解酶、重金属转运/解毒蛋白及一个无特征功能的假定蛋白, 可能与玉米行粒数的发育形成密切相关。
Kernel number per row in maize is a significant trait in determining yield components and it has great significance to study its genetic mechanism. This report studied 80 Jilin maize inbred lines in field experiments at Jilin Changchun and Jilin Meihekou, and measured kernel number per row in 2014 and 2015. At the same time, whole-genome resequencing was performed for the association population using second generation sequencing technology, and the obtained single nucleotide polymorphisms (SNPs) markers were used for subsequent analysis. The results revealed that the range of phenotypic traits of kernel number per row was from 12.0 to 41.6 and the broad-sensed heritability was 70.5% in four environments. A total of 19 SNP markers significantly associated with kernel number per row were detected by a genome-wide association study. Of these, two markers located at bins 2.04 and 3.08 of chromosome frame were detected in the experiments at Changchun and Meihekou in 2015, respectively, and 14 SNP markers located within the quantitative trait loci had been previously mapped. Four candidate genes, such as the genes encoding the receptor for ubiquitination targets protein, metal dependent phosphohydrolase, heavy metal transport/detoxification protein and putative protein with no characteristic function, were identified from the range of linkage disequilibrium of the significant SNP makers and predicted that they were closely associated to the development of the kernel number per row.
玉米的单产主要由百粒重、行粒数、穗行数、单位面积有效穗数等构成, 其中行粒数作为玉米产量的重要组成因素, 不仅其遗传力较高[1], 而且与产量呈显著正相关[2]。因此, 探究玉米行粒数性状的遗传机制, 对于指导玉米高产育种, 提高玉米单产水平具有重要意义。
目前, 研究者们利用AFLP和SSR等分子标记定位了大量控制玉米行粒数的QTL。2005年Lan等[3]以191个F2代单株为试验材料, 利用91个SSR和20个AFLP标记共定位得到了9个控制玉米行粒数的QTL, 解释的表型变异率在5.4%~13.7%之间。2010年Li等[4]利用沈5003和掖178杂交所衍生的210个F2:3家系为作图群体, 利用207个SSR标记在不同的磷处理条件下定位得到11个控制玉米行粒数的QTL。其中位于第5染色体的QTL解释的表型贡献率高达14.35%。2016年Huo等[5]利用2个F2:3家系分别定位得到3个和6个QTL与玉米行粒数密切相关, 它们分布在第1、第2、第3、第7和第10染色体上。其中位于第1号染色体上的数量性状位点qEL1.10在多个穗部性状的定位中都被检测到, 说明该位点是一个控制玉米产量的多效QTL。2016年Chen等[6]利用D276/D72/ A188/Jiao51进行四元杂交所产生的后代群体为供试材料, 利用221个SSR标记共定位得到了6个控制玉米行粒数的QTL, 其中有4个QTL均位于第5染色体上。尽管在上述研究中均定位得到了控制玉米行粒数的染色体区域, 但受所选研究群体和遗传标记密度的制约, 多数定位结果的置信区间比较大, 有效性较低。近年来, 随着植物基因组测序技术的快速发展。第3代分子标记SNP和全基因组关联分析方法得到了越来越多的应用, 为解析玉米复杂性状的遗传构成开辟了新的途径。
本研究选取80份吉林省核心玉米自交系作为关联群体, 使用第2代测序技术对玉米行粒数开展全基因组关联分析。精细定位与行粒数紧密关联的分子标记, 深入挖掘玉米种质资源中控制行粒数的等位基因。为选育行粒数多, 产量高的玉米新品种提供理论支持。
80份吉林省玉米核心自交系由吉林农业大学生物技术中心提供, 于2014年和2015年分别在吉林长春及梅河口进行种植, 采用完全随机实验区组设计, 行长3.00 m, 行距0.65 m, 每小区种植3行, 3次重复, 密度80 000株 hm-2, 田间管理条件与常规生产相同。
在植株生理成熟后, 每小区内随机收取10个果穗, 每穗选取较整齐的一行测定行粒数。使用Microsoft Excel软件计算小区内平均数及对表型性状数据进行描述性统计分析, 使用DPS软件计算变异系数和相关系数并进行方差分析。按照Knapp等[7]提出的公式h2= σ g2/(σ g2+σ e2)计算遗传力, 公式中σ g2为遗传方差, σ e2为环境方差。
采用康为世纪新型植物基因组DNA提取试剂盒提取80份试验材料基因组, 经1%琼脂糖凝胶电泳及Nanodrop1000微量紫外可见分光光度计检测质量, 检测合格的DNA样品使用Illumina测序仪进行测序。质控后测序数据通过BWA软件[8]比对到玉米B73基因组序列RefGen_v3, 比对结果经SAMtools软件[9]去除重复, 同时为保证选取SNP的可信性, 采用贝叶斯模型进行群体SNP的检测与筛选, 以缺失率小于10%、最小等位基因频率大于0.05等阈值为标准, 共得到1 490 007个高质量的SNP标记用于后续分析。
运用MEGA6.0软件(http://www.megasoftware.net/)构建遗传距离矩阵, 群体主成分分析采用GCTA软件(http://cnsgenomics.com/software/gcta/)进行, 利用Admixture软件(http://www.genetics.ucla.edu/software/ admixture/)分析群体遗传结构, 使用PLINK软件[10]计算关联群体的连锁不平衡平均衰减距离(LD)。试验群体的亲缘关系分析借助GAPIT工具[11]进行, 使用FarmCPU模型[12]进行SNP标记与行粒数性状的关联分析, 当SNP标记的P< 0.000 001时, 判断其与研究性状具有显著关联。
根据与玉米行粒数显著关联的SNP标记在玉米基因组中的物理位置, 在玉米遗传学和基因组学数据库(http://www.maizegdb.org/gbrowse)上与玉米B73基因组序列RefGen_v3进行比对, 在LD范围内扫描玉米行粒数候选基因。候选基因的注释及功能预测借助玉米遗传学和基因组学数据库及美国国立生物技术信息中心数据库(https://blast.ncbi.nlm.nih.gov/Blast.cgi)进行。
2014年长春、2014年梅河口、2015年长春、2015年梅河口行粒数平均值分别为27.54、27.04、26.88和26.70, 变异范围为12.0~41.6, 变异幅度明显高于Huo等[5]利用Mo17 × TY6及W138 × TY6构建的2个F2:3家系群体, 这可能是因为自然群体的遗传背景更为丰富, 积累了更多的遗传变异。变异系数及方差分析的结果显示, 本研究选取的各自交系间行粒数性状差异显著。行粒数遗传力为36.4%, 说明玉米行粒数性状受环境影响较大[3]。相关分析显示, 各年份及环境间行粒数数据显著相关(表1)。
| 表1 行粒数数据统计分析 Table 1 Statistics on kernel number per row |
测序得到的高质量数据量为3230.75 Gb, 平均每个样品40.38 Gb, 通过BWA软件比对到参考基因组, 群体样本平均比对率为98.82%, 对基因组的平均测序深度为17.62, 平均覆盖度为88.39%。与其他数据集合如Maize hapmap2比较, 其测序深度等略有不足, 但与参考基因组的相似度、测序深度和覆盖度均达到了重测序分析的要求。
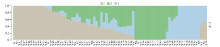
该群体的LD分析结果显示, r2=0.1时关联群体连锁不平衡平均衰减距离为5.2 k。以群体的遗传距离矩阵为基础, 参考主成分分析结果及所选材料的血缘信息, 可将选取的80份玉米自交系材料分为3个亚群, 其中亚群1主要由Reid种质及其改良系构成; 亚群2主要由国内种质构成, 同时还包含少量改良Reid种质和含有热带血缘的种质; 亚群3由Lancaster种质和欧洲种质构成。据此采用Admixture软件以本次研究假定的祖先群体个数为3, 即K=3进行群体遗传结构分析, 分析结果如图1。
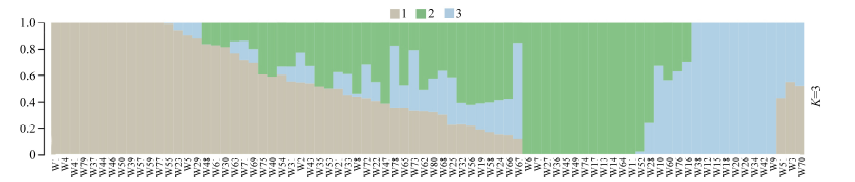 | 图1 群体结构图不同颜色片段的长度表示该个体基因组中某个祖先所占的比例。Fig. 1 Group structure plot The length of the different color segments represents the proportion of an ancestor in the individual genome. |
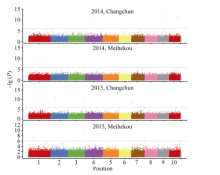
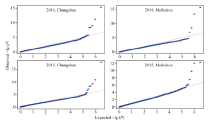
以P< 0.000 001 (-lg P > 6)为标准, 各环境下共检测到19个与玉米行粒数显著关联的SNP标记, 分布于除第5染色体外的各条染色体上。在染色体框1.04中检测到了3个与行粒数显著关联的SNP标记, 染色体框7.01中检测到了2个, 与Tuberosa发现的数量性状遗传位点成簇现象相吻合[13]。位于染色体框3.08的sKNR14标记和位于染色体框2.04的sKNR15标记在2015年长春和梅河口均被检测到, 其中sKNR14在两环境中的表型贡献率分别达到了38.49%和24.95% (表2)。标记sKNR17物理位置位于候选基因GRMZM2G101036内, 为同义突变SNP, 不影响该基因编码蛋白的一级结构(图2与表2)。关联分析中得到的QQ统计图见图3。
| 表2 与玉米行粒数显著相关的SNP标记(P< 0.000001) Table 2 SNPs identified to be associated with kernel number per row (P < 0.000001) |
对与玉米行粒数显著关联SNP标记的基因组区域进行扫描, 得到了4个候选基因(表2)。GRMZM2G101036基因编码的蛋白含有F-box结构域, 功能预测为泛素化目标受体(Receptor for Ubiquitination Targets)。GRMZM5G 835562基因编码一个无特征功能的假定蛋白。GRMZM2G 088397基因编码的蛋白含HD_3结构域, 预测其功能为金属依赖性磷酸水解酶(metal dependent phosphohydrolases)。GRMZM2G313009基因编码的蛋白含一个HMA结构域, 功能预测为重金属转运/解毒蛋白(heavy metal transport/detoxification protein)。
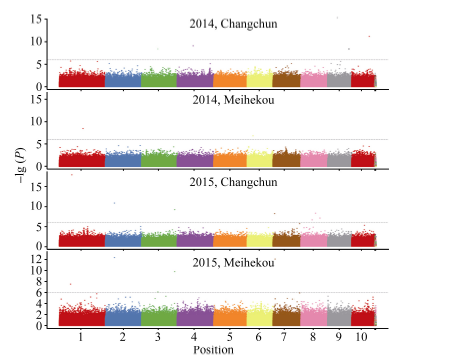 | 图2 全基因组关联分析曼哈顿图Fig. 2 Manhattan plot of genome-wide association study |
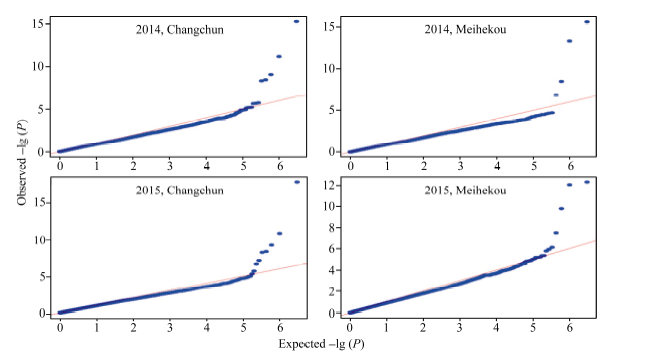 | 图3 全基因组关联分析QQ图Fig. 3 Quantitle-quantitle plot of Genome-wide association study |
玉米是异花授粉作物, 在育种过程中容易受到环境及人为选择的影响, 其LD的衰减较快[14], 适合于应用关
联分析方法进行分析。本实验中共得到19个与玉米行粒数显著关联的SNP标记, 将它们与已有研究结果相比较, 发现14个SNP标记位于已定位到的QTL置信区间内, 如染色体框1.05中定位到的标记sKNR1位于Lu等[15]定位的区间mzetc34-umc1053中; 染色体框9.03中定位到的标记sKNR2位于刘宗华等[16]定位的区间phi065-umc1271中; 标记sKNR3位于染色体框9.06, 与代国丽等[17]定位的SSR标记bnlg1191相近, 物理位置相差4 Mb; 位于染色体框4.05的sKNR5与杨俊品等[18]定位的SSR标记csu74位置接近; 位于染色体框8.03的2个标记sKNR10和sKNR11与杨国虎等[19]通过2个近等基因系群体定位的区间bnlg2082-bnlg2046相吻合; 在2015年长春和梅河口均检测到的标记sKNR14和sKNR15与Huo等[5]定位的区间umc1767-umc2152和umc2032- umc1065一致, 分别位于染色体框3.08和2.04, 说明本试验结果具有很高可信性。但本实验定位到的显著性SNP标记中, sKNR7、sKNR8、sKNR12、sKNR16和sKNR18未找到与之相符的研究结果, 这可能是因为玉米行粒数以多基因遗传为主, 由多个微效基因加性效应决定[1, 20], 而传统定位方法受遗传背景和分析方法的限制, 难以对微效多基因进行定位[18]。
根据显著关联的SNP标记的物理位置, 在连锁不平衡范围内进行扫描, 得到了4个候选基因。其中GRMZM2G101036基因编码的蛋白含有F-box结构域, F-box结构域通常由40~50个氨基酸组成, 是与SCF (Skp1-Cullin-F-box protein)复合体中的Skp1或Skp1类似蛋白结合的区域, 起到调节不同情况下蛋白质间交互作用的功能, 通常通过泛素-蛋白酶体途径(ubiquitin-Proteas pathway)参与细胞的周期调控、转录调控、凋亡或信号转导[21]。
GRMZM2G088397基因编码的蛋白中包含一个HD_3结构域, HD_3结构域是HDc家族的成员, 该家族由Aravind等[22]于1998年首次提出, 并认为具有HDc家族结构域的蛋白是有金属依赖性的磷酸水解酶(metal dependent phosphohydrolases)。Yakunin等[23]的研究表明, 在大肠杆菌核苷酸基转移酶中, HD结构域调控的磷酸水解活性与修复tRNA的3° -CCA末端密切相关。目前还未有研究表明含有HD_3结构域的蛋白在植物体中的作用。
候选基因GRMZM2G313009的核酸序列与拟南芥基因组中一个重金属转运/解毒蛋白家族(heavy metal transport/detoxification superfamily protein)基因直接同源, 且与水稻基因组中一个富脯氨酸表达蛋白(proline-rich protein putative expressed)基因直接同源。其编码的蛋白中含有HMA (heavy metal ATPase)结构域, 已有大量研究表明HMA蛋白是一个能够水解ATP并利用释放的能量驱动重金属离子跨膜转运的蛋白种类, 在Zn、Cd、Pb、Co等重金属离子的运输过程中起重要作用, 其工作机制与钠泵和钾泵类似。植物、细菌和人的HMA氨基酸序列具有高度的同源性[24, 25]。目前已知的HMA家族蛋白均与排出转运及细胞内区室化有关[26]。
候选基因GRMZM5G835562未发现直接同源序列, 也未发现其编码蛋白中有典型结构域, 其功能有待进一步研究。
The authors have declared that no competing interests exist.
作者已声明无竞争性利益关系。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|