

棉花早熟性相关的性状多为复杂的数量性状, 是由多个基因和环境共同作用的结果, 其遗传基础研究比较困难。利用连锁作图和关联分析方法, 对目标性状进行基因定位, 找出与性状相关联的位点, 为解析复杂性状的基因来源提供了新的手段。本文分别以早熟亲本新陆早8号和新陆早10号为母本, 以陆地棉标准系TM-1为父本构建F2作图群体, 利用在亲本间筛选出的多态性SSR引物, 通过JoinMap 3.0软件构建了2张遗传图谱, 以Win QTLCart 2.5复合区间作图法在F2~F2:3中定位到控制全生育期、苗期、蕾期、花铃期和霜前铃数等早熟性相关性状的37个QTL。控制早熟性的有利等位基因多数来自早熟祖先611波和金字棉。多数性状在两类材料中由不同的基因控制, 且以加性遗传为主。在由43个陆地棉品种材料构成的自然群体中通过关联分析检测到54个与这些早熟性相关性状极显著关联的位点。研究表明连锁作图和关联分析的检测结果具有较高的可比性。本研究为早熟陆地棉聚合改良以及分子标记辅助育种打下了基础。
The traits related to early maturity in upland cotton are quantitative traits usually affected by multiple genes and environments, it is difficult to dissect their genetic basis. However, both linkage mapping and association analysis provide new tools toward interpreting the gene sources of complex traits. In this study, two F2mapping populations were constructed by using upland genetic standard line TM-1 as male to cross with female parents Xinluzao 8 and Xinluzao 10, respectively. Thirty-seven QTL for traits related to early maturity were identified by the composite interval mapping (CIM) method of Win QTLCart 2.5 in the F2-F2:3 of the two mapping populations. Positive alleles at these loci were mostly from early maturity ancestor 611-B and King cotton, respectively. In addition, most of the traits were controlled by different genes in the two sets of gene pools, showing primarily additive effects. Furthermore, in a natural population consisting of 43 different Upland cotton cultivars, 54 genetic loci significantly associated with early maturity related traits were detected through genome-wide association analysis, showing great comparability with the linkage mapping results. Our study provided insights into the genetic bases of early maturity and laid a foundation for early maturity gene pyramiding and marker-assisted selection of upland cotton.
新疆是目前我国棉花生产的主产区, 种植面积占全国棉花面积的一半以上, 由于受生育期有效积温偏低、无霜期短等气候条件的制约, 中、晚熟棉花品种难以正常成熟吐絮, 因此早熟性是该区棉花育种的首要目标。经典的数量遗传学研究表明, 陆地棉早熟性是由多基因控制的数量性状, 其遗传方式中除存在显性[1,2,3]、加性效应外[4,5,6,7,8,9,10,11], 还存在上位性效应[12], 与生育阶段、开花时期、第一果枝节位、吐絮速率、霜前花率等性状有关[13,14,15,16], 且与产量、品质间存在显著的遗传负相关。
随着分子标记技术的不断发展和完善, 研究人员通过分析QTL (quantitative trait locus)数目和效应来研究复杂数量性状的遗传机理, 定位控 制数量性状的基因位点, 在分子水平上剖析复 杂的数量性状。迄今为止, 利用棉花数据库(http:// www.cottonmarker.org/)公布的微卫星(simple sequence repeat, SSR)标记已经构建了多张高密度的棉花种间[17,18,19,20,21]及种内[22,23,24,25,26,27,28]遗传图谱, 为研究棉花复杂数量性状的遗传机制和基因资源的发掘利用奠定了基础。目前, 国内外学者对棉花产量、品质、抗性等许多重要性状进行了QTL定位研究[29,30,31,32,33], 有关早熟性及其相关性状的基因定位研究报道较少[34,35,36,37,38]。范术丽等[35]利用中棉所36×TM-1组合, 定位了果枝始节、现蕾期、开花期、全生育期、霜前花率等控制熟性相关性状的12个QTL; 张先亮等[34]定位了控制生育期性状的5个主效QTL。Guo等[36]定位了位于染色体16、21、25上的控制第一果枝节位的3个QTL。努斯来提等[37]分别定位了位于染色体5和LG02上的控制第一果枝节位、始节高度和蕾期的5个QTL。Li等[38]定位了控制第一果枝节位和始节高度的14个QTL, 分别位于染色体A1、A5、A6、A9、A11、A13、D3、D6、A12/D12和LG1上。
新陆早8号、新陆早10号是具有不同遗传背景的新疆棉区种植的早熟陆地棉品种, 全生育期均为120 d左右, 高产优质、抗病性好。本研究以新陆早8号、新陆早10号为母本, 分别与陆地棉遗传标准系TM-1配制杂交组合, 利用两组合的F2及F2:3分离群体对苗期、现蕾期、花铃期、霜前铃数和全生育期等早熟性相关性状进行QTL定位研究; 同时利用另外收集的43份早熟陆地棉品种材料(主要来自金字棉及前苏联衍生系)构成的自然群体进行关联分析, 探讨了早熟性相关性状的分子遗传机理及其基因来源。以期为进一步挖掘与早熟性状相关联的优异等位变异及早熟陆地棉分子标记辅助育种打下基础。
新陆早8号、新陆早10号均由新疆生产建设兵团八师农业科学研究所育成, 是20世纪90年代末北疆早熟陆地棉区域种植的主栽品种, 其中新陆早8号是前苏联品种611波衍生系, 而新陆早10号则为金字棉衍生系。用于关联分析的早熟陆地棉材料(表1)主要来源于新疆生产建设兵团七师、八师农业科学研究所及中国农业科学院棉花研究所。2006年冬季在海南岛分别以新陆早8号、新陆早10号作母本、以陆地棉遗传标准系TM-1作父本配制杂交组合。2007年在南京农业大学江浦试验站种植两组合的亲本、F1及43份陆地棉材料构成的关联分析群体, 并做自交留种。采用育苗移栽, 随机区组排列, 单行区, 重复2次。行长5.0 m, 行距0.8 m, 每行15株。2008年在八师农业科学研究所种植两作图群体的亲本、F2及关联分析群体, 2009年种植两群体的亲本、F2:3家系及关联分析群体。均采取完全随机区组设计, 重复2次。采用55 cm (40+35+40)宽膜覆盖, 3行区, 行长5.0 m, 株距16 cm, 家系和关联分析群体材料每行均种植约30株。田间管理同一般大田管理。
2007、2008和2009年分别在南京农业大学江浦试验站和新疆建设兵团八师农业科学研究所调查了分离群体的亲本、F1、F2和F2:3家系及关联分析群体的早熟性相关性状, 即出苗后, 从中行选择5株(江浦)或12株(八师)壮苗挂牌定株, 调查各株的出苗、现蕾、开花、吐絮等时期; 在收获期调查各株霜前吐絮铃数, 即霜前铃数(pre-frost bolls)。根据不同生育时期计算各生育期, 其中苗期(seedling stage)为棉花出苗至现蕾天数; 蕾期(budding stage)为棉花现蕾至开花天数; 花铃期(flowering and bolling stage)为棉花开花至吐絮天数; 全生育期(growth stage)为棉花播种至吐絮天数。取性状均值用于数据分析。
以本实验室构建的饱和海陆种间遗传图谱为基础, 每隔一定距离(10 cM)选择一个标记, 共选取覆盖棉花全基因组的402对SSR标记引物对关联分析群体进行标记基因型分析。将SSR标记扩增出的条带量化, 同一位点处等位基因的出现和不出现分别记为“1”和“0”, 用于关联分析。另用7 331对SSR及EST-SSR引物对新陆早8号、新陆早10号2个作图群体的亲本进行多态性筛选, 分别选出亲本间呈现多态性的引物191和108对; 用这些多态性引物对2个F2分离群体进行标记基因型分析, 以阿拉伯数字“1”记为母本P1带型、“2”为父本P2带型、“3”为F1杂合带型。用JoinMap 3.0[39]分析标记间的连锁, 构建分子遗传图谱。作图函数为Kosambi函数[40]。 依据本实验室框架图[41,42,43]将连锁群定位到相应染色体上。
|
|
表1 用于关联分析的43份陆地棉材料的系谱来源Table 1 Pedigree of 43 Upland cotton cultivars selected for association analysis |
|
|
续表1 用于关联分析的43份陆地棉材料的系谱来源Table 1 (Continuous) Pedigree of 43 Upland cotton cultivars selected for association analysis |
采用WinQTLcart 2.5的复合区间作图法[44](composite interval mapping)进行早熟相关性状的QTL定位。性状的LOD值阈值经1000次排列测验确定。LOD值3.0以上表示显著性QTL, LOD值2.0~3.0表示可能性QTL。由曲线峰顶向两侧各下降1和2个LOD值来确定QTL的90%和95%的置信区间。采用水稻上常用的QTL的命名方法[45], 以字母“ q”开头表示QTL, 后接性状名称的缩写, 再接染色体或连锁群的编号, 最后是该染色体上控制此性状的QTL序号。
利用PowerMarker 3.25[46]分析关联分析群体遗传多样性, 获得等位变异数目、等位基因频率、基因型数目以及基因多样性等参数值。应用Structure 2.2[47,48], 对所选棉花品种进行基于数学模型的类群划分, 并计算材料相应的 Q值。利用TASSEL[49]软件包计算连锁不平衡(linkage disequilibrium, LD)配对检测的矩阵图, 使用其广义线性模型(general linear model, GLM)程序, 将各个体的 Q值作为协变量分别对标记变异和每个性状的表型变异进行回归分析。
在新陆早8号×TM-1组合(Pop1)的F2~F2:3中, 检测到早熟性相关性状QTL14个, 其中显著性QTL 8个(表2)。在新陆早10号×TM-1组合(Pop2)的F2~F2:3中, 检测到23个QTL, 其中显著性QTL 13个(表3)。
2.1.1 全生育期 在Pop1两个分离世代中检测到控制生育期的2个显著性QTL和3个可能性QTL (表2), 分别位于染色体A2、D8、D12和连锁群LG01上, LOD值为2.53~3.83, 解释表型变异的4.0%~ 7.0%。缩短生育期的等位基因均来自早熟亲本新陆早8号, 共缩短生育期8.7 d。其中QTL q-GS-A2-1和 q-GS-LG01的早熟等位基因分别缩短生育期2.8 d和1.8 d, 其显性效应分别缩短生育期2.1 d和0.9 d, 而QTL q-GS-D12-1的早熟等位基因缩短生育期0.7 d, 但其显性效应增加生育期2.0 d。在Pop2两个分离世代中检测到3个显著性QTL和5个可能性QTL (表3), 分别位于染色体A5、A6、A7/D7、D1、D7、D8、D9上, LOD值2.64~3.58, 解释表型变异的4.3%~12.0%。减少生育期的多数有利等位基因来自于早熟亲本新陆早10号, 缩短生育期0.8~3.9 d, 但这些QTL的显性效应能延长生育期0.6~5.5 d, 对早熟性不利。
2.1.2 苗期 在Pop1的F2:3家系中检测到控制苗期的显著性QTL和可能性QTL各1个(表2), 分别位于LG03和A2上, LOD值分别为3.04和2.70, 解释表型变异的6.0%和5.0%。缩短苗期的等位基因均来自晚熟亲本TM-1, 分别缩短苗期0.76 d和0.96 d。其中QTL q-SS-A2-1的显性效应延长苗期0.24 d。在Pop2的F2:3家系中检测到控制苗期的4个显著性QTL和1个可能性QTL (表3), 分别位于A6、D1和D8上, LOD值2.82~3.90, 可解释表型变异的7.0%~10.0%。缩短苗期的等位基因均来自早熟亲本新陆早10号, 能缩短出苗期0.81~1.16 d。QTL q-SS- A6-1的显性效应能缩短苗期0.24 d, 其余QTL的显性效应均延长苗期。
2.1.3 蕾期 在Pop1的F2群体中分别检测到控制蕾期的2个显著性QTL (表2), 均位于连锁群LG02上, LOD值分别为5.98和3.43, 分别解释表型变异的9.0%和10.0%。缩短蕾期的等位基因分别来自于TM-1和新陆早8号, 缩短蕾期1.23 d和1.33 d, 但其显性效应分别增加蕾期0.66 d和0.42 d。在Pop2的F2群体中检测到控制蕾期的2个显著性QTL (表3), 分别位于D7、D8上, LOD值为7.27和3.15, 解释表型变异的16.0%和5.0%。缩短蕾期的等位基因均来自早熟亲本新陆早10号, 分别能缩短蕾期1.24 d和0.63 d。其中QTL q-BS-D8-1的显性效应能缩短蕾期0.42 d。
2.1.4 花铃期 在Pop1的F2~F2:3群体中检测到控制花铃期的显著性QTL和可能性QTL各1个(表2), 分别位于A2和D9上, LOD值分别为3.69和2.65, 解释表型变异的7.0%和4.0%。缩短花铃期的等位基因均来自于早熟亲本新陆早8号, 分别缩短花铃期2.68 d和1.23 d。这2个QTL的显性效应分别能缩短花铃期1.89 d和0.31 d。在Pop2的F2~F2:3群体中检测到控制花铃期的2个显著性QTL和4个可能性QTL (表3), 分别位于A6、A6/D6、A7/D7、D7、D13上, LOD值2.62~4.54, 可解释表型变异的4.0%~ 12.0%。缩短花铃期的多数有利等位基因来自于早熟亲本新陆早10号, 缩短花铃期1.15~2.41 d, 但其显性效应均延长花铃期。
2.1.5 霜前铃数 在Pop1的F2~F2:3群体中共检测到控制霜前铃数的2个显著性QTL和1个可能性QTL (表2), 位于D1、D2、D7染色体上, LOD值为2.82~5.82, 解释表型变异的6.0%~9.0%。增加霜前铃数的等位基因均来自晚熟亲本TM-1, 分别增加霜前铃数0.04~1.52个。其中QTL q-PFB-D1-1的显性效应增加霜前铃数1.12个。在Pop2的F2群体中检测到控制霜前铃数的2个显著性QTL (表3), 位于D7、A6/D6上, LOD值分别为8.82和4.49, 可解释表型变异的16.0%和7.0%。增加霜前铃数的等位基因均来自晚熟亲本TM-1, 分别增加霜前铃数1.74个和0.92个。其中QTL q-PFB-A6/D6-1显性效应可增加霜前铃数0.92个。
| 表2 新陆早8号×TM-1组合中检测到的相关性状QTLTable 2 QTL related to early maturity detected in the cross of Xinluzao 8×TM-1 |
| 表3 新陆早10×TM-1组合中检测到的相关性状QTLTable 3 QTL related to early maturity detected in the cross of Xinluzao10×TM-1 |
用挑选的402对SSR引物, 对43份早熟棉花品种进行多态性检测, 共筛选到174对多态性引物(43.28%), 检测到486个差异条带。不同的SSR引物在43个品种间检测到的差异条带数不同, 变幅为2~7个, 其中92对引物各检测到2个差异条带, 占差异条带数的52.29%; 57对引物各检测到3个差异条带, 占32.75 %; 引物NAU5467、NAU5468、NAU5472和NAU5486分别检测到7个差异条带, 位于D2、D5、D9和A9染色体上, 位点多态信息含量(polymorphism information content, PIC)变幅为0.15~0.76。所选早熟陆地棉品种基因多样性指数平均为0.40, 变幅为0.04~0.78; 平均PIC值为0.33。以上结果表明, 所选的SSR引物检测的差异条带数目和基因多样性的跨度较大, 但平均值较低, 虽说明了研究材料在基因组水平上变异比较丰富, 但也反映了陆地棉遗传基础的狭窄性。
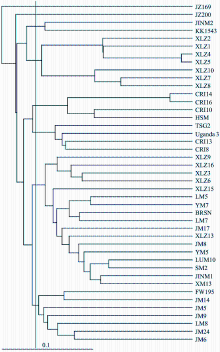 | 图1 棉花品种的UPGMA聚类树系图品种代号同表1。Cultivars’ names are the same as those listed in Table 1.Fig. 1 Nei-UPGA dendrogram of 43 cotton cultivars |
根据174个SSR标记的Nei’s遗传距离聚类, 在相似性系数0.03水平上大致将43个早熟棉花品种(系)分为8类(图1)。其中, 与贝尔斯诺有亲缘关系的新疆棉区育成的品种新陆早3号、新陆早9号、新陆早15、新陆早16和具有斯字棉、金字棉、乌干达棉血统的辽棉5号、辽棉7号、豫棉5号、豫棉7号、锦棉1号、晋棉17、鲁棉10号和湘棉13等17个品种聚为一类。这些品种反映了不同陆地棉系统之间相互交叉的现象。5个晋棉品种、辽棉8号和汾无195等7个品种聚为一类, 这些品种均是金字棉衍生的早熟类型。塔什干2号、乌干达3号、中棉所8号和中棉所13等4个品种聚为一类, 这些品种分别是前苏联、金字棉衍生系。黑山棉、中棉所10号、中棉所14、中棉所16等4个品种聚为一类, 其中中棉所10号是从黑山棉中经过系统选择育成, 中棉所14、中棉所16是以中棉所10号为共同亲本杂交育成的, 均为黑山棉衍生系。新陆早1号、新陆早2号、新陆早4号、新陆早5号、新陆早7号、新陆早8号、新陆早10号等7个品种聚为一类, 其中新陆早1号、新陆早2号、新陆早8号来源于前苏联品种611波, 新陆早4号来源于司42, 新陆早5号来源于岱字棉15, 新陆早7号来源于塔什干2号, 而新陆早10号来源于金字棉。这些品种均为新疆棉区育成的早熟品种, 反映了新疆早期和近代早熟陆地棉育种中不同系统之间的融合现象。来自乌兹别克斯坦的品种KK1543和金字棉衍生的品种锦棉2号聚为一类。晋中169和晋中200各自划分为一个类群, 这两个品种是分别从朝阳1号及窝及1号中通过定向选择育成。
利用3年2点4环境数据及各环境平均值分别进行关联分析, 共检测到与5个早熟性相关性状显著关联( P<0.01)的位点54个, 分布在除A10和D13以外的所有24条染色体上, 其中有21个位点同时与2个以上的性状显著关联(表4)。
|
| 表4 与早熟性相关性状相关联的位点及其解释的表型变异Table 4 Loci associated with traits related to maturity and the phenotypic variation explained |
|
| 续表4 与早熟性相关性状相关联的位点及其解释的表型变异Table 4 (Continuous) Loci associated with traits related to maturity and the phenotypic variation explained |
与全生育期显著关联的位点共19个, 其中位于A9、A11和D6上的3个位点BNL1317、NAU5428和NAU2687可在4个环境中同时被检测到, 位于A2、D1、D4和D12上的7个位点分别在2~3个环境中同时被检测到, 可解释表型变异的9.64%~ 13.55%。与苗期显著关联的位点共有15个, 位于A2、A3、A11、D1、D11和D12上的6个位点分别在2个以上环境中同时被检测到, 可解释表型变异的9.36%~17.73%。与蕾期显著关联的位点共有18个, 位于A2、A5、A7、A8、A9、D2、D5、D10和D11上的9个位点分别在2个以上环境中同时被检测到, 可解释表型变异的9.57%~27.99%。与花铃期显著关联的位点共有15个, 其中位于A9、D4、D6上的3个位点BNL1317、NAU4058和NAU2687可在4个环境中同时被检测到, 位于A11、D7、D9上的3个位点分别在2~3个环境中同时被检测到, 可解释表型变异的10.94%~28.07%。与霜前铃数显著关联的位点共有15个, 其中位于染色体D6、D8、D10、D11和D12上的6个位点可在2~3个环境中同时被检测到, 可解释表型变异的12.05%~26.34%。
一些位点与多个早熟性状相关联, 如位于染色体D2上的位点NAU5467同时与全生育期、苗期、蕾期和花铃期显著关联, 在染色体D7上的位点JESPR297同时与全生育期、苗期和花铃期显著关联。说明这些位点的有利等位基因的表达可能是组成性的, 在不同的生育阶段均发挥作用。
基于DNA标记对棉花种质资源的遗传多样性分析, 可获得丰富的遗传信息, 为种质改良和新品种选育提供依据。本研究利用基本覆盖棉花基因组的分子标记对早熟种质资源进行分析, 除晋中169和晋中200与其他品种间的亲缘关系较远外, 多数品种集中在金字棉系谱中, 亲本来源单一。反映出所选早熟陆地棉的亲缘关系较近、遗传多样性较低。根据品种系谱可以看出, 分子聚类虽然倾向于将具有共同亲本或遗传基础相近的品种聚在一起, 但没有真正从来源上将品种区分开。究其原因, 主要是所选品种相对较少, 多态性低。多态性标记在不同染色体上的分布不均, 试验获取的PIC偏低, 也导致聚类结果难以同实际系谱相吻合。另外, 不同早熟棉育种单位之间材料交流比较广泛, 基因的交换、重组及渐渗造成遗传背景较为复杂, 使得聚类分析结果难以与系谱衍生系分类信息相对应。相对于系谱分析, 分子聚类结果对于陆地棉早熟育种的亲本选配具有更大的指导作用。
陆地棉早熟性是复杂的数量性状, 其遗传方式也较为复杂, 采用经典数量遗传学研究方法得出的结论不尽一致, 显性[5,6,7]、加性[8,10,11,12,13,14,15]、超显性和上位性[16]遗传均有报道。本研究在Pop1中检测到控制全生育期的5个QTL, 来自新陆早8号的有利等位基因共缩短全生育期8.68 d, 显性效应共增加全生育期0.20 d。检测到的蕾期和花铃期的6个QTL中, 来自新陆早8号的有利等位基因分别减少蕾期和花铃期1.33 d、3.91 d, 其显性效应则延长蕾期1.08 d、缩短花铃期2.20 d。在关联分析中也检测到能缩短生育期且与新陆早8号相关联的等位变异。在Pop2中检测到控制全生育期的8个QTL中, 来自新陆早10号的等位基因共缩短生育期11.86 d, 但显性效应共增加生育期25.61 d。检测到的苗期、蕾期和花铃期的13个QTL中, 来自新陆早10号的有利等位基因共缩短苗期、蕾期、花铃期4.24、1.87和7.46 d, 而其显性效应分别延长苗期4.12 d、缩短蕾期0.35 d、增加花铃期12.47 d。以上结果表明, 新陆早8号组合的早熟性以加性效应为主, 新陆早10号组合中控制苗期、蕾期的早熟基因也以加性遗传为主, 而 控制花铃期和全生育期的QTL显性效应高于加性效应。
两个组合中除D8上控制全生育期的QTL所在的位置相同外, 其他性状QTL的位置均不同。由于新陆早8号和新陆早10号的早熟基因分别来自611波和金字棉, 说明早熟性在这2个早熟祖先系列中是由不同的基因控制的。
与传统的QTL作图方法相比, 关联分析省去了亲本间多态性筛选、作图群体基因型的鉴定等繁琐工作, 直接利用软件处理表型数据与SSR等位变异, 其QTL定位不依赖图谱, 方法简单直观。然而该方法的缺陷是不能估计QTL的具体位置及加性和上位性效应。从本研究的多年多点关联分析结果看, 有54个位点与早熟性相关的5个性状呈极显著关联, 而两作图群体的连锁分析中共检测到与熟性相关的37个QTL。由于关联分析群体的早熟来源大部分与连锁分析亲本的早熟来源相同(表1), 2种分析方法检测到较多一致性QTL。例如两种分析方法均检测到位于染色体A2、A5、A6、D1、D7和D12上的控制棉花全生育期的QTL; 位于A2、A6和D1上控制苗期的QTL; 位于D7、D8上控制蕾期的QTL; 位于A2、A6、A7、D6、D7和D9上控制花铃期的QTL以及位于D6、D7上控制霜前铃数的QTL。这些相对应的QTL在本实验室构建的参考图谱上的位置除个别稍有偏离外, 大部分极为接近。经分析发现, 关联分析中检测到的早熟性相关性状的多数优异等位变异来源于金字棉衍生系育成的品种, 如新陆早10号、晋棉14等。在Pop2中也检测到较多与熟性有关的QTL, 其有利等位基因也来自新陆早10号。说明两种方法的定位结果在一定程度上可以相互验证。金字棉是早熟陆地棉改良的骨干亲本, 与新陆早8号的祖先亲本611波相比, 早熟性状更为突出。这表明具有金字棉遗传背景的早熟亲本携带较多的早熟优异等位基因, 对早熟性相关性状的贡献率较大。这些来自于早熟亲本相关性状的基因位点, 可用于早熟棉分子标记辅助育种。
构建了2张早熟陆地棉遗传图谱, 在F2~F2:3中定位了全生育期、苗期、蕾期、花铃期和霜前铃数等熟性性状的37个QTL, 控制早熟性的有利等位基因多来自早熟祖先611波和金字棉。多数性状在这两种遗传背景中由不同的基因控制, 且以加性遗传为主; 自然群体中呈现多态的174对引物揭示出品种基因多样性指数为0.40, 平均PIC值为0.33, 说明这些材料在基因组水平上虽存在比较丰富的变异, 但遗传基础仍较狭窄; 根据分子标记检测结果聚类, 将43份早熟棉种质划分为8类, 反映出早期和近代早熟棉育种中不同血统之间的交叉、融合现象; 关联分析检测到54个与早熟性相关性状极显著关联的位点。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
 3.0 and 22 suggestive QTLs, 3.0 > LOD
3.0 and 22 suggestive QTLs, 3.0 > LOD