

{kind=link}
{kind=link}
{kind=link}
{kind=link}
玉米-大豆轮作及氮肥施用对土壤细菌群落结构的影响
[周岚1, 2, ** , 杨永1, ** , 王占海3 , 陈阜1 , 曾昭海1, *  ]
]
]
|
|


* 通讯作者(Corresponding author): 曾昭海, E-mail:zengzhaohai@cau.edu.cn
**同等贡献(Contributed equally to this work)
在大豆开花期分别对3个施氮水平下(0、50和100 kg hm-2)大豆连作(大豆-大豆-大豆)、玉米-大豆轮作I (大豆-玉米-大豆)及玉米-大豆轮作II (玉米-玉米-大豆), 应用PCR-DGGE技术研究玉米-大豆轮作及施氮对土壤细菌群落结构变化的影响。结果表明, 随着施氮水平的提高, 3种种植方式土壤中细菌群落多样性、丰富度均呈减少趋势。高氮处理(100 kg hm-2)明显降低了大豆连作、玉米-大豆轮作II根际土壤细菌群落多样性及丰富度, 玉米-大豆轮作I根际土壤细菌群落多样性及丰富度略有降低。玉米-大豆轮作I种植方式可减轻氮肥对其根际细菌群落多样性和丰富度的影响, 但施氮明显改变了其细菌群落结构。玉米-大豆轮作II中大豆根际土壤细菌群落结构较为稳定, 受氮肥影响较小。在3种种植方式的土壤中, 分布着酸杆菌门、变形菌门及厚壁菌门细菌, 其中前两门菌群占主导地位。
In soybean flowering period, took rhizosphere soil samples from three treatments including continuous cropping soybean (soybean-soybean-soybean), maize-soybean rotation I (soybean-maize-soybean) and maize-soybean rotation II (maize- maize-soybean). Each treatment had three levels of nitrogen application including 0, 50, and 100 kg ha-1. Soil microbial community changes under maize-soybean rotation system and various nitrogen applications were investigated by the techniques of denaturing gradient gel electrophoresis (DGGE) analysis. The results showed that the biological diversity and abundance of three kinds of soil microbial community had a decline trend along with the increase of nitrogen application levels. Under high nitrogen application level (100 kg ha-1), a significant decrease in diversity and abundance of rhizosphere soil microbial communities was observed in both continuous cropping soybean system and maize-soybean rotation II system. And in maize-soybean rotation I system, there was only a slightly decrease in diversity and abundance of rhizosphere soil microbial communities. Therefore, 1-year maize in rotation with soybean (soybean-maize-soybean) can alleviate the effect of nitrogen fertilizer on rhizosphere soil microbial diversity and richness, but the effect of nitrogen application significantly changed its bacterial community structure. Maize-soybean rotation II system (maize-maize-soybean) was less affected by nitrogen fertilizer and showed relatively high stability. In addition, in those three kinds of soil, there were the categories of Acidobacteria, Proteobacteria and Firmicutes, in which, Acidobacteria and Proteobacteria were predominant.
土壤微生物是农田生态系统的重要组成部分, 对于土壤中物质转化和循环具有重要调节作用, 是指示土壤环境变化, 反映土壤质量的最重要指标之一[1]。前人依靠传统方法仅能获得土壤微生物总数的0.1%~1.0%[2,3], 可能与土壤原位菌群存在较大差异[4]。变性梯度凝胶电泳(denaturing gradient gel electrophoresis, DGGE)是使用一对特异性引物扩增从样品中提取的DNA或rRNA, 得到长度相同但序列有异的16S rDNA产物, 然后在添加一定浓度线性梯度变性剂的聚丙烯酰胺凝胶上电泳分离。Muyzer等[5]1993年首先将DGGE技术应用于分子微生物生态学研究。与传统平板培养方法相比, PCR-DGGE技术可以检测到土壤中大量不可培养的细菌, 能够更精确地反映土壤微生物多样性, 比传统培养方法在代表性上具有巨大优势, 在细菌群落多样性研究中被广泛应用。如李梓正等[6]应用PCR-DGGE技术研究不同退化草地上细菌群落多样性的变化; Qiao等[7]应用PCR-DGGE技术研究了燕麦和箭筈豌豆混播对细菌群落结构的影响; 刘恩科等[8]应用PCR-DGGE技术研究了不同施肥及种植制度对细菌群落结构的影响。
豆科作物与禾本科作物轮作是传统农业的精华, 一方面豆科作物的生物固氮可以为禾本科作物提供氮素营养, 另一方面禾本科作物收获后创造的土壤环境又有利于促进与之轮作的豆科作物的生物固氮[9]。东 北平原是我国玉米、大豆主产区, 也是重要的商品粮基地。玉米与大豆轮作可以解决大豆连作障碍问题, 增加大豆产量, 改善大豆品质, 维持土壤健康, 缓解大豆生产中病虫草害[10,11,12]。大豆根际微生物被认为是造成大豆连作障碍的重要因子之一[13], 研究玉米大豆轮作条件下大豆根际细菌群落多样性与结构的变化可以进一步明确轮作效应的作用机制。本文应用PCR-DGGE技术分别对大豆连作、玉米-大豆轮作I (大豆-玉米-大豆)及玉米-大豆轮作II (玉米-玉米-大豆)在3个施氮水平下(0、50和100 kg hm-2)的大豆根际细菌群落进行了研究, 明确轮作效应与土壤细菌群落的关系, 为建立合理轮作模式及氮肥管理提供参考依据。
2010年在吉林农业科技学院试验基地进行盆栽试验。作物有效生长季为125~135 d, 生长季≥10℃有效积温为2450~2600℃, 全年日照时数2300~2500 h, 全年平均降水量为650~750 mm。盆栽用塑料桶高50 cm, 上口直径40 cm, 下口直径30 cm。供试大豆品种为黑农51。供试土壤为沙壤土, 试验开始时供试土壤0~25 cm耕层含有机质14.30 g kg-1、全氮1.85 g kg-1、全磷1.53 g kg-1、全钾26.70 g kg-1、碱解氮117.40 mg kg-1、速效磷95.90 mg kg-1、速效钾93.30 mg kg-1, pH 6.9。
采用随机区组双因素试验设计, 因素分别为土壤种类和施氮水平。土壤分别来自大豆连作田(大豆-大豆)(T1)、大豆-玉米轮作田(大豆-玉米)(T2)和玉米连作田(玉米-玉米)(T3); 施氮水平分别为0、50和100 kg hm-2。每个处理4个重复, 共36盆。将盆底钻孔, 将盆埋在土壤中, 模拟大田种植。每盆装10 kg风干土, 播种前按P2O5105 kg hm-2、K2O 120 kg hm-2的磷、钾肥均匀施入土壤, 保证肥料与土壤充分混匀。挑选饱满均匀的大豆种子, 每盆播5粒, 并接种大豆根瘤菌剂, 出苗后间苗留3株。在大豆开花期, 在大豆根系周围去除表层土, 采集2~10 cm耕层, 将4个重复土样混为一个样品, 立即放入冰盒中带回实验室, 于-20℃冰箱中保存, 待提取各样品DNA。
1.3.1 土壤总DNA的提取 称取0.3 g土样, 按照超快型土壤DNA提取试剂盒(购自华越洋生物科技有限公司)说明书提取各土壤样品总DNA。
1.3.2 PCR扩增 PCR扩增16S rDNA V3高变区, 引物分别为带有GC夹子的357f-GC (5'-CGCCCG CCGCGCGCGGCGGGCGGGGCGGGGGCACGGGGGGCCTACGGGAGGCAGCAG-3')和517R (5'-ATT ACCGCGGCTGCTGG-3')[8]。PCR扩增体系为50 μL, 其中10×buffer 5 μL, dNTP(2.4 mmol L-1) 4 μL, 上下游 引物(10 pmol μL-1)各1 μL, Taq DNA聚合酶(2.5 U μL-1) 0.5 μL, ddH2O 37.5 μL。引物由上海生工生物工程有限公司合成, ddH2O自备, 其他试剂均购自宝生物工程(大连)有限公司。PCR扩增程序为预变性95 ℃ 3 min, 然后30个循环为94 ℃ 30 s, 59 ℃ 30 s, 72 ℃ 1 min 10 s, 最后72 ℃延伸10 min, 扩增长度约为230 bp。利用1.5%的琼脂糖凝胶电泳, 溴化乙锭染色, 检测PCR扩增产物。
1.3.3 DGGE 按DNA片段纯化试剂盒(北京三博远志生物技术有限公司)说明书将细菌16S rDNA的PCR产物6管浓缩至约30 μL, 取2 μL用超微量分光光度计检测, 浓度约为300 ng μL-1, 4℃保存。取13 μL上述浓缩后的样品加入7 μL loading buffer进行DGGE电泳。DGGE凝胶梯度为6%~12%, 变性剂梯度为25%~55%, 温度60℃, 在100 V电压下电泳10 h。
1.4.1 微生物群落的多样性、丰富度和均匀度 利用DGGE图谱的数字化结果计算多样性指数 H (Shannon-weaver index)、丰富度 S 和均匀度 E 。
多样性指数 Pi = ni / N , 式中, ni 为每个带的亮度, N 为某一样品所有带亮度的和[7]。
Shannon-Weaver index = - 
均匀度 E = H /ln S , 式中, H 为Shannon-Weaver index, S 为丰富度即某一样品的条带数[7]。
戴斯系数: Cs = 2 j /( c + d ), 式中 j 是样品C和D共有的条带, c 和 d 分别是样品C和D中各自的条带数[8]。
1.4.2 微生物群落结构的分析 采用SAS 9.0软件根据不同处理DGGE图谱的条带亮度和位置的数字化数值对微生物群落结构进行主成分分析。
取得了各样品16S rDNA V3区PCR产物很好的分离效果(图1)。从图谱可以很直观地看出, 不同处理细菌DGGE指纹图谱在条带数量、亮度及位置等方面均存在明显差异。图谱中各泳道条带的数量及亮度可表征不同样品中细菌的种类和数量。9种处理在DGGE图谱中条带数量(丰富度S)由19至31不等(表1), 其中大豆连作土壤在不施氮肥时, 条带数最多为31个, 条带数较多(25个以上)的处理还有T1-50、T2-0、T2-50、T3-0、T3-50; 3种种植方式的大豆根际土壤中, 随施氮量的增加, 条带数量均呈递减趋势(表1)。因此, 在不施氮或施氮水平较低时, 土壤中细菌丰富度较高, 施加氮肥可减少土壤中细菌丰富度。在3个施氮水平下, 大豆连作土壤细菌丰富度均较高, 玉米-大豆轮作II土壤细菌丰富度较低, 玉米-大豆轮作I土壤丰富度在0 kg hm-2和50 kg hm-2氮处理下均处于中间水平, 而在高氮处理下最高。根据图谱中条带的数量及亮度, 由Bio-Rad 4.6.2 quantity one软件计算了各处理的细菌群落多样性指数, 其变化同丰富度变化类似。随施氮水平的提高, 各处理土壤中细菌群落的多样性指数均呈减少趋势。高氮肥处理明显降低了大豆连作土壤及玉米-大豆轮作II土壤的细菌多样性指数, 而玉米-大豆轮作I减缓了氮肥带来的影响。不同处理对细菌群落的均匀度影响较小。
 | 图1 细菌DGGE指纹图谱T1: 大豆-大豆-大豆连作; T2: 大豆-玉米-大豆轮作; T3: 玉米-玉米-大豆轮作。3个施氮水平0、50和100 kg hm-2。Fig. 1 DGGE pattern of bacterial communityT1: soybean-soybean-soybean continuous cropping; T2: soybean-maize-soybean rotation; T3: maize-maize-soybean rotation.Three leaves of N are 0, 50, and 100 kg hm-2. |
| 表1 轮作及施氮对细菌多样性指数、丰富度及均匀度的影响 Table 1 Effect of rotation and nitrogen application on bacterial Shannon-Weaver index, richness, and evenness |
由图2可知, 轮作及氮肥均对样品细菌群落结构产生了一定的影响。不施氮肥处理T1-0、T2-0和T3-0分别位于第四、一、三象限, 表明不施氮肥条件下, 不同种植方式对土壤中细菌群落结构产生了较大的影响; T2-50、T3-50位于第二象限, T1-50位于第四象限, 表明在低氮肥处理下, 轮作土壤中细菌群落结构更为接近, 与连作土壤细菌群落结构差异较大; T1-100、T2-100均位于第三象限, T3-100位于第二象限, 表明在高氮肥处理下, 轮作I和连作大豆根际更为接近, 而与轮作II差别较大。T1-0、T1-50较为接近, 与T1-100差别较大, 表明高氮肥处理对大豆连作土壤细菌群落结构影响较大, 其差异主要分布在横轴上。T2-0、T2-50和T2-100差别较大, 其差异主要分布在纵轴上; T3-0、T3-50、T3-100较为接近, 表明施氮仅是减少了玉米-大豆轮作II中大豆根际土壤细菌多样性及丰富度, 但对细菌群落结构影响较小。
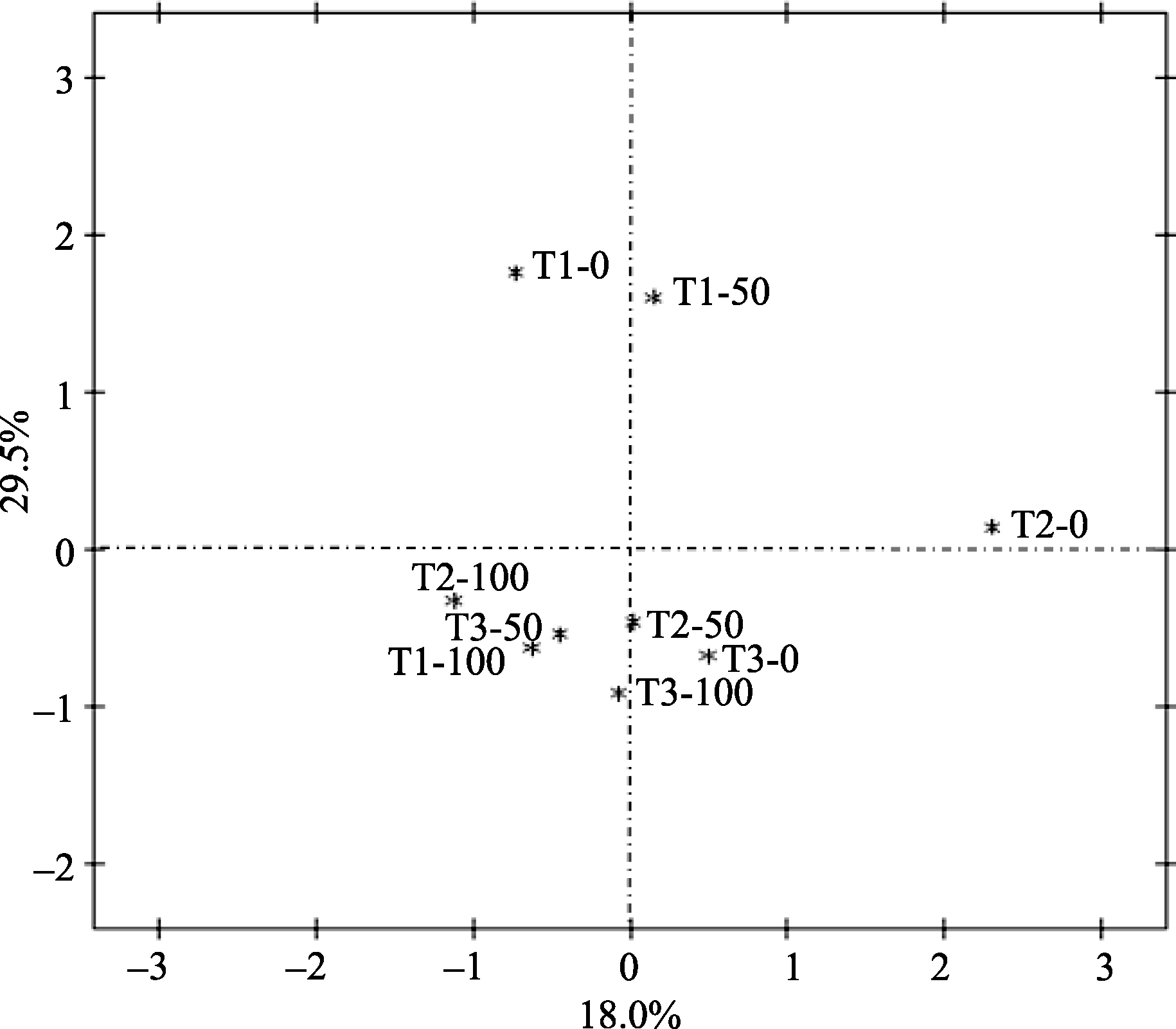 | 图2 细菌群落结构主成分分析T1: 大豆-大豆-大豆连作; T2: 大豆-玉米-大豆轮作; T3: 玉米-玉米-大豆轮作。3个施氮水平为0、50和100 kg hm-2。Fig. 2 PCA analysis of bacterial community compositionT1 oybean-soybean-soybean continuous cropping; T2: soybean-maize-soybean rotation; T3: maize-maize-soybean rotation. Three leaves of N are 0, 50, and 100 kg hm-2. |
根据戴斯系数, 可进一步对不同处理间的相似度进行数字化分析。由表2可知, 在3个施氮水平下, 大豆连作(T1)同玉米-大豆轮作II (T3)的相似度分别为50.8、56.1和55.2, 同玉米-大豆轮作I (T2)的相似度分别为53.6、54.6和68.2, 表明根际土壤中细菌群落与所种植作物有着密切关系。在T1和T2处理中, 大豆的参与度较高, 因此两者更为相似。T1-0同T1-50、T1-100的相似度分别为72.6、48.4, T1-50同T1-100的相似度为50.6; T2-0同T2-50、T2-100的相似度分别为62.9、52.1, T2-50同T2-100的相似度为62.8; T3-0同T3-50、T3-100的相似度分别为66.1、60.0, T3-50同T3-100的相似度为69.7, 表明不同土壤均表现出低施氮处理与不施氮处理更为相似, 氮肥的影响随其量的增加而逐渐加强, 高氮肥处理与不施氮处理差别较大。
| 表2 不同处理的戴斯系数 Table 2 Dice coefficient of different treatments (%) |
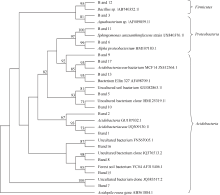
从DGGE图谱中挑取了15个条带, 利用去除GC夹子的引物重新PCR扩增, 克隆后测序, 序列长度在169~194 bp之间。利用Blast软件将测序得到的基因序列与GenBank数据库进行序列比对, 获取近似菌株的16S rDNA序列。其中有5个克隆(band5、band7、band8、band10和band16)序列同不可培养的细菌较为相似, 相似性在98%~100%之间; 其他克隆均与可培养的细菌具有较高的相似性, 相似性在93%~100%之间(表3)。利用Mega5.1中邻接法(Neighbor-Joining)将挑取的15个克隆序列与15个相近菌株序列建立16S rDNA系统发育树如图3所示, 共分为三大门类, 即酸杆菌门、变形菌门和厚壁菌门。
| 表3 测序结果 Table 3 Results of sequencing |
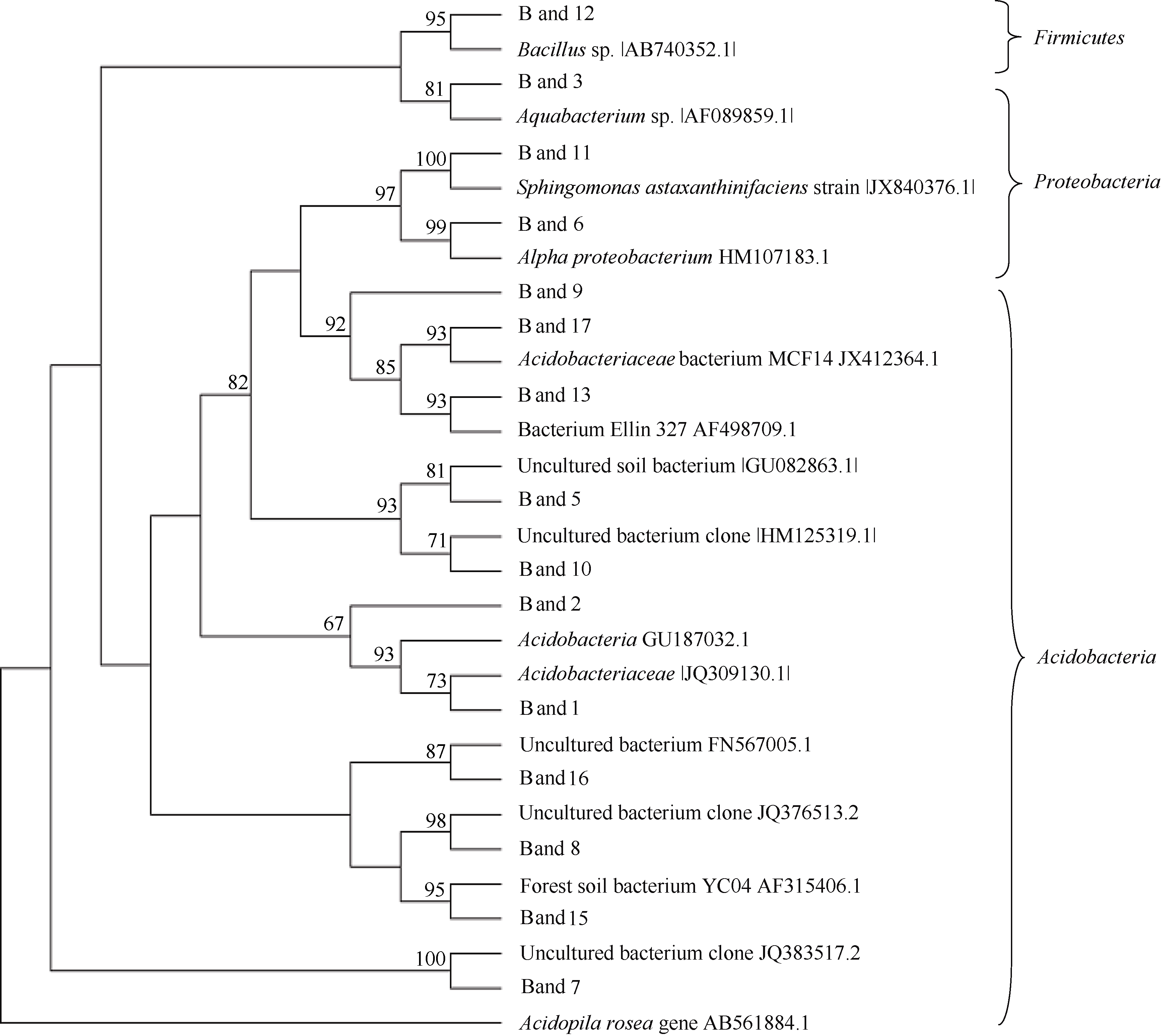 | 图3 采用N-J法建立系统发育树Fig. 3 Phylogenetic of 16S rDNA sequences by Neighbor-joining method |
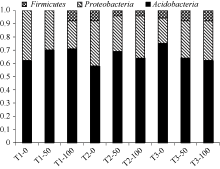
酸杆菌和变形菌广泛分布于作物根际及自然界土壤中, 是土壤菌群结构的两大重要类群[14]。由图4可知, 样品中细菌多属于酸杆菌门和变形菌门, 分别占总量的58%~75%和19%~38%; 厚壁菌门所占比例较少, 甚至在连作大豆根际土壤不施氮和施氮50 kg hm-2处理中未被发现。在两种轮作土壤中不同施氮水平下均有酸杆菌门、变形菌门、厚壁菌门分布。
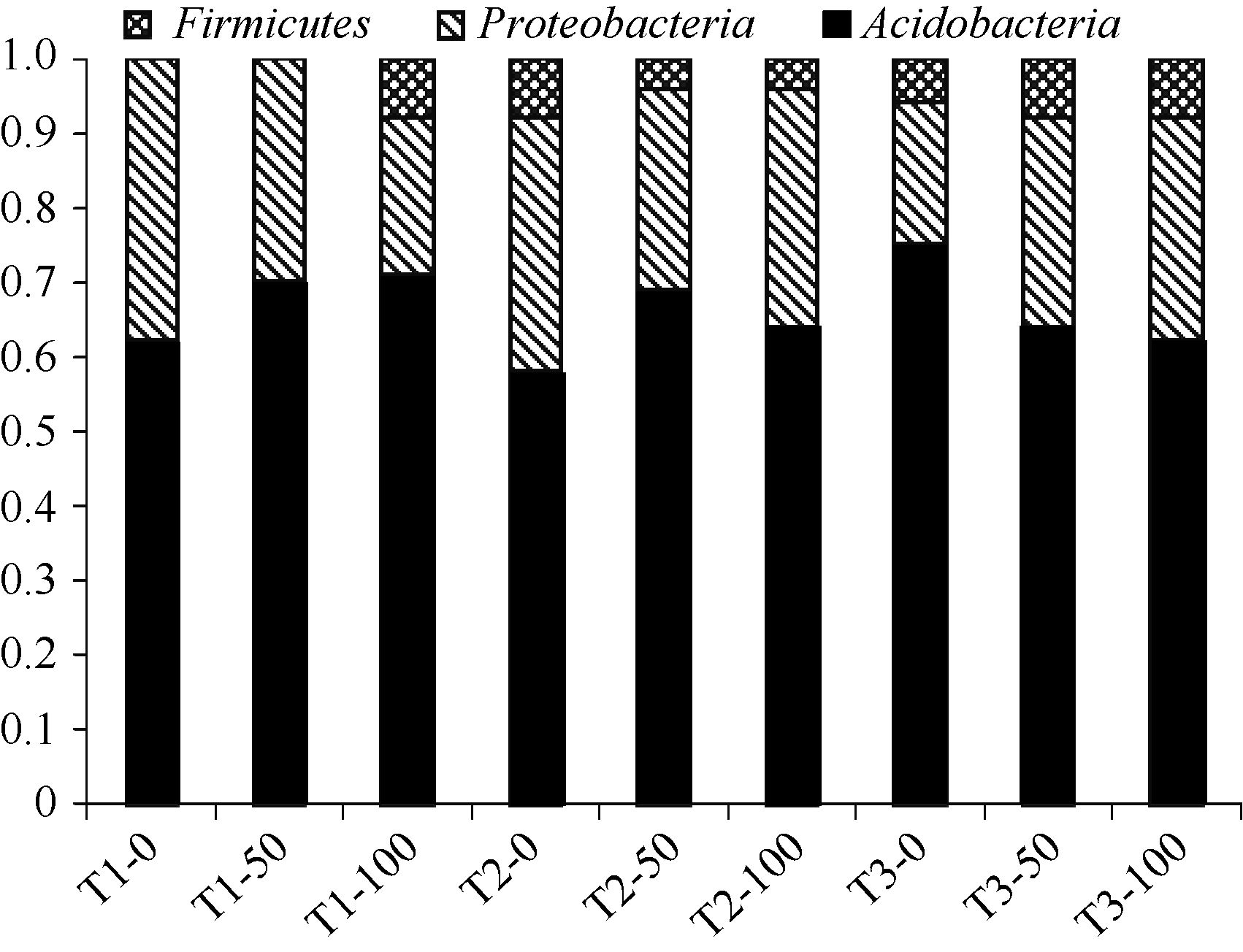 | 图4 不同处理细菌群落组成的比较Fig. 4 Comparison of bacterial community in different treatments |
随施氮水平的增加, 大豆连作、玉米-大豆轮作I及玉米-大豆轮作II土壤中细菌群落多样性、丰富度均呈减少趋势, 与王光华等[15]研究结果一致, 氮肥减少了土壤中细菌群落多样性及丰富度。施氮量50 kg hm-2与不施氮处理细菌群落多样性及丰富度差别较小, 而施氮量100 kg hm-2明显降低了土壤细菌群落多样性及丰富度。高氮处理(T1-100)下大豆连作土壤细菌群落结构与低氮(T1-50)、不施氮处理(T1-0)具有较大差异; 施氮处理(T2-50、T2-100)的玉米-大豆轮作I土壤细菌群落结构较为接近, 与不施氮处理(T2-0)差别相对较大。细菌群落的多样性、丰富度对外界响应较为一致, 但与细菌群落结构是否改变并不统一。本研究中氮肥亦仅降低了玉米-大豆轮作II土壤中细菌多样性及丰富度, 对其细菌群落结构影响较小, 与时鹏等[16]研究结果一致。
马春梅等[17]研究显示, 相对于玉米, 大豆对微生物的繁衍具有更好的促进作用。在不施氮和低氮水平下, 细菌群落多样性及丰富度依次为大豆连作土壤>玉米-大豆轮作I土壤>玉米-大豆轮作II土壤。与玉米-大豆轮作II土壤相比, 玉米-大豆轮作I土壤细菌群落与大豆连作土壤相似度更高。这可能与大豆根际细菌群落更为丰富有关。在高氮水平下, 玉米-大豆轮作I土壤细菌多样性及丰富度最高, 表明大豆-玉米隔年轮作可减缓高氮肥对作物根际细菌的影响; 相对于大豆连作和玉米-大豆轮作II土壤, 玉米-大豆轮作I提高了细菌群落多样性及丰富度, 与吴凤芝等[18]和姚钦[19]研究结果一致。轮作II土壤中在不同施氮水平下细菌群落结构最为稳定。经条带回收测序发现, 3种种植方式大豆根际土壤中包含酸杆菌门、变形菌门和厚壁菌门三大门类菌群, 其中酸杆菌门和变形菌门细菌所占比例较大, 与张丽等[20]对农田细菌群落组成的研究结果类似。
高氮肥处理明显降低了大豆连作及玉米-大豆轮作II土壤细菌多样性、丰富度, 而玉米-大豆轮作I减缓了高氮处理对细菌丰富度和多样性带来的影响; 氮肥对细菌群落的影响随施氮量增加逐渐增强, 高施氮量(100 kg hm-2)与不施氮处理差别最大。高氮处理明显影响了大豆连作和轮作I土壤细菌群落结构, 但对轮作II土壤细菌群落结构影响相对较小。不同处理细菌群落的均匀度基本无差异。3种土壤中主要包含酸杆菌门、变形菌门和厚壁菌门细菌, 且前两门细菌占主导地位。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|