
{kind=link}
{kind=link}
玉米穗行数全基因组关联分析
张焕欣 , 翁建峰
, 翁建峰* , 张晓聪, 刘昌林, 雍洪军, 郝转芳, 李新海*
, 翁建峰, 张晓聪, 刘昌林, 雍洪军, 郝转芳, 李新海
|
|


* 通讯作者(Corresponding authors): 翁建峰, E-mail:jfweng@126.com; 李新海, E-mail:lixinhai@caas.cn
第一作者联系方式: E-mail:zhanghuanxin150@163.com
穗行数是玉米产量的重要组成性状, 其遗传解析对高产育种具有指导意义。本文以203份主要玉米自交系为材料, 2007年在新疆乌鲁木齐、吉林公主岭和海南三亚进行穗行数测定; 采用分布于玉米基因组的41 101个单核苷酸多态性(SNP)标记对穗行数进行关联分析。共鉴定出9个与穗行数显著关联(
, WENG Jian-Feng, ZHANG Xiao-Cong, LIU Chang-Lin, YONG Hong-Jun, HAO Zhuan-Fang, LI Xin-Hai
Kernel row number (KRN) is one of grain yield components in maize (
玉米穗行数(kernel row number, KRN)形成于小穗分化期, 由小穗成对分生组织数目决定[ 1]。穗行数是决定玉米产量的主要构成因素, 属于数量性状, 广义遗传力较高[ 2], 其遗传解析对玉米高产育种具有指导意义。分子标记的发展使得QTL作图成为解析穗行数遗传结构的有效方法[ 3]。目前, 关于穗行数定位研究报道较多, 影响穗行数的QTL在玉米10条染色体上均有分布。Ma等[ 4]利用综3×87-1构建的294份重组自交系(recombinant inbred line, RIL)群体检测出13个穗行数QTL, 分别位于第1、第3、第4、第5、第8、第9和第10染色体。Lu等[ 5]利用掖478×丹340的150个F2:3家系共定位到13个控制穗行数的QTL, 位于染色体框7.03位点来自丹340的穗行数主效QTL qkrn7可解释平均表型变异17.86%。Guo等[ 6]用郑58×昌7-2的231个F2:3家系在两种播种密度下进行穗行数QTL定位, 分别检测到8个和7个穗行数QTL。谭巍巍等[ 7]利用掖478×黄早四与齐319×黄早四构建的2套F2:3家系在染色体框5.04、6.05-6.07、7.02和10.03鉴定出穗行数QTL。江培顺等[ 8]采用元分析方法整合了35篇文献中定位的184个分布于玉米10条染色体上的穗行数QTL, 共发掘出除位于第6染色体外的22个Meta- QTL。
研究者利用不同的遗传群体和QTL作图方法挖掘出诸多穗行数QTL, 但是, 受标记密度的限制, 检测到的QTL置信区间较大、有效性较低, 而且当控制该性状的基因在两亲本之间相同时则不能被检测到, 难以提供育种群体中优良等位基因的信息。与传统双亲QTL作图相比, 全基因组关联分析(genome-wide association study, GWAS)具有高分辨率、高通量的优势, 有利于鉴定现有种质资源中的有利等位基因[ 9, 10]。Huang等[ 11]对950个水稻品种进行GWAS, 分别检测出32个和10个与开花期和产量性状显著关联的遗传位点。Weng等[ 12]利用41 101个SNP对284个玉米自交系进行株高全基因组关联分析, 鉴定出105个遗传位点。借助关联分析与连锁作图, 将抗玉米丝黑穗病主效位点 qHS2.09精细定位至约1 Mb[ 13]。Tian等[ 14]、Buckler等[ 15]和Brown 等[ 16]以含5000个RIL的玉米巢式作图群体的全基因组分析, 鉴定出影响叶形、开花期和花序性状的遗传位点及候选基因。
迄今, 有关玉米穗行数的全基因组关联分析及相关候选基因发掘的研究报道较少。本文采用203份玉米自交系组成的关联作图群体进行全基因组分析, 挖掘玉米种质中蕴含的穗行数优异等位基因, 为克隆控制玉米产量性状基因奠定基础。
203份玉米自交系, 由中国农业科学院作物科学研究所提供。2007年, 将其分别种植于新疆乌鲁木齐(XJ)、吉林公主岭(JL)和海南三亚(HN)。采用行长4.5 m, 行距0.6 m, 每行20株的完全随机区组试验设计, 2次重复。按照常规生产条件进行田间管理。收获后, 每个小区随机取10个果穗调查穗行数, 平均值用于统计分析。
采用改良CTAB法[ 17]提取203份自交系幼叶基因组DNA, 用0.8%琼脂糖凝胶电泳和NanoDrop 2000分光光度计检测DNA质量, 合格的样品DNA用于SNP分型。本文采用Illumina公司开发的MaizeSNP50 BeadChip芯片, 该芯片包括56 110个SNP标记, 均匀覆盖玉米自交系B73全基因组。SNP基因型检测参照Illumina公司提供的操作指南, 采用Illumina BeadStation 500 G SNP分型系统完成。芯片基因型数据质量控制参考Weng等[ 12], 采用 41 101个高质量的SNP标记进行关联分析。
采用SAS 9.1.3 (SAS Inc., Cary, NC)的PROC UNIVARIATE、PROC GLM和PROC CORR程序分别进行描述性统计量分析、方差分析和相关分析。按照Knapp等[ 18]提出的公式 h2 = σg2/( σg2 + σgl2/ n + σe2/ nr)估算遗传力, 其中 σg2为遗传方差, σgl2为基因型与环境互作的方差, σe2为误差方差, n为地点数, r为重复数。
利用软件STRUCTURE V2.3[ 19]检测该群体的遗传结构, 以5000个最小等位基因频率大于0.2且在染色体均匀分布的SNP标记进行估计, Weng等[ 12]将该群体划分为旅大红骨、四平头、Lancaster、PB、BSSS和PA 6个亚群。通过软件SPAGeDi[ 20], 采用相同的5000个SNP估计自交系间的亲缘关系。
运用软件TASSEL V3.0[ 21]中的混合线性模型(mixed-linear model, MLM)程序, 在考虑研究材料群体结构与亲缘关系情况下, 进行SNP标记与穗行数的关联分析。在 P< 0.0001水平上, 判定SNP标记与穗行数关联的显著性。
收集1994—2013年定位的穗行数QTL信息; 借助玉米基因组数据库MaizeGDB确定其物理位置。若GWAS检测到的SNP在物理位置上与前人QTL定位结果重叠, 则认为二者具有一致性。
基于GWAS结果, 利用与穗行数显著相关SNP的60 bp源序列, 比对B73的参考序列RefGen_v2 (MGSC) (http://blast.maizegdb.org/home.php?A=LAST_UI)。依据Weng等[ 12]研究结果, 本文关联群体平均LD衰减距离为27.7 kb, 在30.0 kb窗口范围内扫描穗行数候选基因。
新疆、吉林和海南3个环境中的穗行数均值分别为13.29、13.86和13.61, 变异系数分别为13.87%、13.84%和14.67%, 说明不同自交系间穗行数的差异较大。穗行数遗传力为81.07%, 与Lu等[ 5]的结果相近。Pearson 相关分析表明, 3个环境中的穗行数数据显著相关(表1)。
| 表1 203份自交系穗行数统计分析 Table 1 Statistics on kernel row number evaluated on 203 inbred lines in three environments |
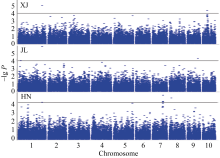
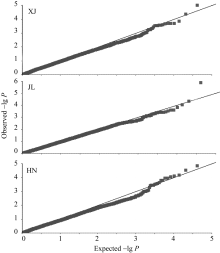
采用TASSEL V3.0软件MLM模型程序, 对203份玉米自交系进行穗行数的全基因组关联分析。 3个环境下共检测到9个与穗行数显著关联( P< 0.0001)的SNP, 分别位于玉米染色体框1.02、1.10、7.03、8.02、9.06和10.03, 其中, 染色体框1.10和7.03分别检测出2个和3个显著性SNP, 最高可解释15.00%的表型变异率(表2)。位于第1染色体框1.10的PZE101220014在新疆和吉林2个环境均被检测到(表2和图1), 而在海南环境中检测到的SNP PZE-101219999与其相差约6 kb。通过TASSEL V3.0软件, 获得3个环境下穗行数全基因组关联分析的QQ图(图2), 关联群体的群体结构得到了较好控制。
 | 图1 穗行数全基因组关联分析XJ、JL和HN分别代表新疆、吉林和海南。黑色水平线代表全基因组关联分析的显著阈值。Fig. 1 Genome-wide association study of kernel row number with mixed linear modelXJ, JL, and HN indicate experiments at Xinjiang, Jilin and Hainan, respectively. Black horizontal line indicates the genome-wide significance threshold. |
| 表2 与穗行数显著关联的SNP位点( P< 0.0001) Table 2 SNPs identified to be associated with kernel row number ( P< 0.0001) |
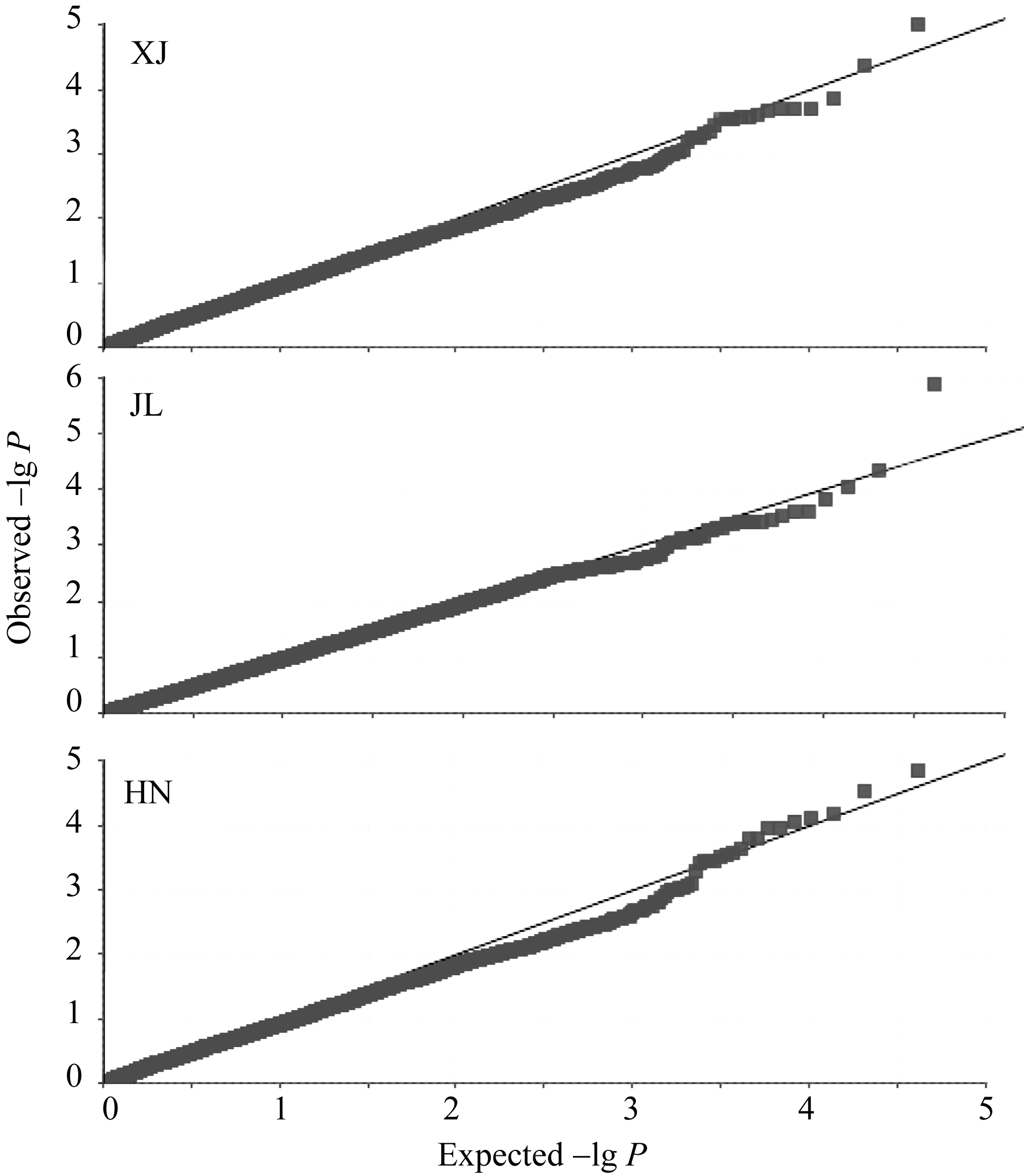 | 图2 3个环境下全基因组关联分析的QQ图横轴表示经过负的常数对数转换的期望 P值, 纵轴表示经过负的常数对数转换观察到的 P值。Fig. 2 Quantile-quantile plot for kernel row number in three environmentsThe horizontal axis shows -lg transformed expected P-values, and the vertical axis shows -lg transformed observed P-values. |
对9个与穗行数显著关联( P< 0.0001)的SNP标记位点进行扫描分析, 获得4个候选基因(表2)。 GRMZM2G398848编码产物是一个含F-box结构域的生长素受体蛋白(auxin signaling F-box containing protein)。 GRMZM2G017087 ( knotted1, kn1)是玉米同源异型盒基因家族成员, 最早发现于玉米顶端分生组织中[ 23], 参与生物发育调控过程。 GRMZM2G 069146编码一个含AP2结构域的蛋白(AP2 domain containing protein)。 GRMZM2G151567的编码产物是富亮氨酸重复的跨膜蛋白激酶(leucine-rich repeat transmembrane protein kinase)。
GWAS是一种以连锁不平衡为基础, 利用分布于全基因组的SNP, 借助统计学工具分析某一群体内目标性状遗传变异的方法。目前, GWAS已应用于鉴定植物病害、开花期、籽粒性状、株高等QTL[ 9, 10, 11, 12, 15, 24, 25, 26], 但玉米穗行数的GWAS研究较少。本文采用203份玉米自交系组成的关联作图群体进行全基因组分析, 在3个环境中共检测出9个与穗行数显著关联( P< 0.0001)的SNP。Massman等[ 27]认为距离小于5 cM的显著SNP可以归为一个QTL, 本文在染色体框1.10检测到的PZE-101219999和PZE-101220014物理距离相距约6.5 kb, 换算后遗传距离小于5 cM, 因此, PZE-101219999和PZE- 101220014应位于同一个QTL。该QTL在3个环境中被重复检测到。连锁不平衡是不同基因座位上等位基因的非随机组合关系[ 28]。关联分析中, 常用 r2度量位点之间连锁不平衡的程度, 当 r2≥ 0.8时认为两位点不独立, 即处于连锁不平衡状态[ 27]。利用HAPLOVIEW V4.0[ 29], SYN17065与PZE-107084987的 r2为0.14, PZE-107084987与PZE-107085390的 r2为0.33, 因此位于染色体框7.03的这3个与穗行数显著关联的标记是独立位点。
连锁定位和关联分析都可以检测数量性状位点, 两种方法定位到的QTL在位置上大部分具有一致性[ 14, 16, 30]。本文检测到的9个SNP中, 有8个位于已定位的QTL区段内(表2)。PZE-101030429位于第1染色体框1.02, 与Guo等[ 6]的研究结果一致。Ma等[ 4]利用综3×87-1构建的294份重组自交系在染色体框1.10的bnlg1597-umc2149处定位到穗行数QTL, 本研究检测到的SNP PZE-101219999和PZE-101220014也位于该区间两标记的物理位置内。位于第8染色体框8.02的PZE-108018598与Liu等[ 22]的研究结果吻合。染色体框7.03共有3个标记与穗行数显著关联, SYN17065、PZE-107084987与PZE-107085390均位于Lu等[ 5]定位的bnlg339- umc1865区间。PZE-110033446位于第10染色体框10.03位置, 与Lu、谭巍巍等[ 5, 7]结果一致。但是本研究发现并非所有GWAS检测到的显著位点(如PZE-109090136)都与已定位的QTL重叠, 原因可能是前人采用的双亲QTL作图法受亲本种质背景和检测微效QTL功效较低的影响。这表明采用全基因组关联分析策略是一种解析穗行数遗传结构的有效方法。
Aranzana等[ 31]对95个拟南芥品种进行GWAS, 发现几个开花期和病原基因均被检测到。Huang等[ 32]利用3 600 000个SNP标记对517个水稻品种开展全基因组分析, 发现6个SNP位点分别与已知基因 qSW5、 GS3、 ALK、 Waxy、 OsC1和 Rc临近。本文对检测出的9个与穗行数显著关联( P< 0.0001)的SNP位点进行扫描分析, 得到4个候选基因。 GRMZM2G398848编码一个含F-box结构域的生长素受体蛋白。F-box蛋白TIR1是生长素的受体, 通过介导一系列生长素反应来参与植物生长发育的调控[ 33]。PZE-101219999位于 GRMZM2G017087( kn1)内部, PZE-101220014位于 kn1上游约6 kb处。 kn1可能维持分生组织细胞的自我更新和控制分枝原基分生组织属性的建成, 玉米突变体 kn1的雄穗分枝数和小穗数均减少, 雌穗小穗对数降低且多数小花不育[ 1, 34]。 GRMZM2G069146编码一个含AP2结构域的蛋白。 IDS1是AP2基因家族成员, 参与调控玉米小穗分生组织的有限性, ids1突变体的每个小穗产生额外的小花[ 35]; IDS1的横向同源基因 SID1与小花分生组织启动及小穗调控有关[ 36]; 水稻基因 SNB与 OsIDS1分别是 ids1和 sid1的直系同源基因, 在控制花序结构与花序分生组织建成等方面具有重要作用[ 37]。 GRMZM2G151567编码一个富亮氨酸重复的蛋白激酶, td1同样编码一个富亮氨酸重复的蛋白激酶, 玉米突变体 td1的雄穗小穗密度、雄蕊数目增加, 雌穗的籽粒行数增加[ 38]; fea2编码一个富亮氨酸的受体样蛋白, 可能通过控制花序分生组织的大小来调控玉米穗行数[ 39, 40]。上述基因信息可能为克隆玉米产量性状相关候选基因提供一定基础。
利用41 101个SNP标记对203份玉米自交系进行穗行数全基因组关联分析, 发现9个与穗行数显著关联( P< 0.0001)的SNP。在显著SNP位点LD区域内发掘出4个候选基因, 分别为 GRMZM2G398848、 GRMZM2G017087、 GRMZM2G069146和 GRMZM2G 151567。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|