


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
烟草表达抗病基因同源物(RGA)的鉴定及RGA-SSR标记的开发
[袁清华, 谢锐鸿, 张振臣, 马柱文, 李集勤, 李淑玲, 陈俊标*  ]
]
]
|
|


烟草是研究植物与病原菌互作的理想材料。鉴定烟草抗病基因及其同源物对揭示抗病机制具有重要意义。近年来公共数据库不断增长的EST序列为烟草表达RGA的鉴定提供丰富的数据。本研究通过拼接GenBank收录的412 325条烟草EST序列, 获得149 606条Uni-EST序列。随后利用已克隆的112个植物
Tobacco is an ideal experimental system for studing on plant-pathogen interaction. Identification of tobacco
烟草是我国乃至世界重要的经济作物。农业生产中, 烟草青枯病、花叶病毒病和黑胫病等严重影响烟叶产量和品质。据全国烟草病虫害预测预报及综合防治网统计, 2010年和2011年, 全国16个烤烟主产省烟草病虫害发生面积达80万公顷, 产量损失6万吨, 产值损失7亿元。因此, 防治烟草病虫害对烟叶生产具有重要作用, 而烟草抗病基因( R基因)和抗病基因同源物(RGA)的克隆则对揭示烟草抗病机制以及制定正确的病虫害防治措施具有重要的意义。
植物 R基因对病原菌无毒基因编码蛋白的识别起关键作用[ 1]。近年来, 通过图位克隆或转座子标签法克隆的植物 R基因超过100个[ 2, 3, 4, 5](http://prgdb. crg.eu/wiki/Species_with_ R-genes)。尽管植物 R基因克服的病原物种类不同, 其编码的氨基酸序列却存在类似结构域, 如核苷酸结合位点(nucleotide binding site, NBS)、富含亮氨酸重复序列(leucine-rich repeats, LRR)、丝氨酸/苏氨酸激酶(serine-threonine kinase, STK)、亮氨酸拉链结构(leucine zippers, LZ)、跨膜结构域(transmembrane domain, TM)、Toll白介素-1区域(toll-interleukin-1 region, TIR)等[ 6, 7, 8, 9]。这些保守结构域为 R基因和RGA的快速鉴定克隆提供捷径。
根据氨基酸序列保守结构域可将抗病基因分为6大类[ 2]。第1类含有NBS和LRR结构域(NBS- LRR)。基于N端是否缺失TIR, 此类 R基因又可分为2个亚类, 即TIR-NBS-LRR和non-TIR-BS-LRR, 例如抗烟草花叶病毒的N基因含有TIR-NBS-LRR结构域[ 7, 10], 而抗丁香假单胞杆菌( Pseudomonas syringae)的拟南芥 Rps2基因则含有CC-NBS-LRR结构域[ 11]。第2类含有LRR和PK结构域(LRR-PK), 例如拟南芥 Fls2和水稻 Xa21基因[ 12, 13]。第3类含有一个胞外LRR结构域, 例如拟南芥 RPP27基因[ 14]。第4类只含有PK结构域, 例如番茄 Pto和香瓜 At1基因[ 15, 16]。第5类包含具有不同抗性机制的其他 R基因, 如玉米 Hm1和大麦 Mlo基因[ 3, 17]。
早期RGA的分离主要通过PCR扩增 R基因保守结构域, 此法已成功从拟南芥[ 18, 19]、大豆[ 20]、水稻[ 21]、玉米[ 22]、小麦[ 23]、烟草[ 24, 25]和其他植物[ 26, 27, 28]中分离出RGA。Bertioli等[ 29]根据NBS设计简并引物, 从花生栽培种和野生种中分离到78个RGA。Gao等[ 25]基于NBS和PK结构域从烟草品种 Nicotiana repanda分离出100个RGA。
与PCR扩增相比, 采用数据挖掘方法从基因组中鉴定RGA效率更高, 基于已公布的植物全基因组序列, 迄今已成功从拟南芥[ 10]、水稻[ 30]、蒺藜苜蓿[ 31]和百脉根[ 32]基因组中鉴定大量RGA。Meyers等[ 10]从拟南芥基因组中鉴定出149个NBS-LRR编码基因和58个其他相关基因。Ameline-Torregrosa等[ 31]从蒺藜苜蓿全基因组草图中鉴定出333个非冗余NBS- LRR, 并预测全基因组含有400~500个NBS-LRR基因。近期, Li等[ 32]从百脉根基因组中成功分离出158个NBS序列。
由于植物基因组中存在大量丧失生物学功能的假基因, 而表达基因占植物基因组比例不到1%, 根据基因组DNA序列开发的RGA多为不表达的假基因[ 32], 严重干扰 R基因的克隆。因此, 鉴定具有表达活性的RGA对 R基因的克隆显得尤为重要。近期, 通过数据挖掘已从植物EST序列中鉴定出系列RGAs。Liu等[ 33]利用454技术测序获得169万条菜豆EST, 随后从中鉴定出364个RGA。Liu等[ 34]利用54个植物R基因氨基酸序列扫描GenBank收录的花生EST序列, 成功鉴定出385个表达花生RGAs。
RGA鉴定后常被用于开发RGA分子标记。基于RGA开发的分子标记主要有RFLP[ 35]、STS[ 36]、SSCP[ 37]、CAPS[ 38]和SSR[ 34]标记等。Sanz等[ 35]根据燕麦RGA序列设计31个RFLP探针, 并成功将53个RGA-RFLP标记定位到燕麦六倍体遗传图谱上( Avena byzantina cv. Kanota × Avena sativa cv. Rollo)。近期, Liu等[ 34]从25个花生RGA中开发出28个SSR标记, 并将其中一个标记RGA121定位到连锁群AhIV上。由于SSR标记具有共显性、多态性高、操作简单且重复性好等优点, 与其他标记相比, 从RGA中开发SSR更有利于抗病基因的定位与克隆。
目前公共核酸数据库GenBank收录的烟草EST序列已达412 325条(截至2012年7月20日), 几乎涵盖不同时期, 不同组织表达的基因, 为烟草表达RGA鉴定提供丰富的资源。鉴于此, 本研究拟利用数据挖掘方法从烟草EST序列中鉴定表达RGA, 继而开发RGA-SSR标记。这将为烟草 R基因的定位与克隆奠定基础。
普通烟草品种24个(表1), 其他烟草种6个, 种植于广东省农业科学院试验农场, 于生长期取叶片提取基因组DNA。
采用TIGR基因指数聚类工具(TIGR gene indices clustering tools, 简称TGICL,
| 表1 供试烟草栽培品种 Table 1 Varieties and wild species of tobacco used in this study |
利用112个已克隆的植物 R基因(表2)氨基酸序列扫描Uni-EST, 从中鉴定表达RGAs。序列比对采用tBlastn工具, 比对分值大于或等于100, 且E值小于或等于1E-10的Uni-EST被认为是候选烟草RGAs (NtRGAs)。
利用Blast工具将NtRGA通过序列比对定位在本塞姆氏烟草( Nicotiana benthamiana)全基因组草图上(http://solgenomics.net/organism/Nicotiana_benthamiana/ nome)。根据比对结果选择比对分值最高、E值最小, 且比对分值大于50, E值小于1E-10的基因组序列作为RGA定位区段。
采用Perl脚本MIcroSAtellite (MISA, http://pgrc. ipk-gatersleben.de/misa/)从RGA序列搜索SSR位点。设置以下SSR搜索限制因子: 最短15 bp, 二核苷酸基序最少重复8次, 三核苷酸基序至少重复5次, 四五六核苷酸基序至少重复4次, 不包含单核苷酸类型。基于SSR侧翼序列, 采用Primer Premier 5软件设计引物。新引物随后用于检测24个烟草栽培种和6个野生种的多态性。
截至2012年6月7日, GenBank收录的烟草属EST序列共412 325条, 其中 N. tabacum334 384条, N. benthamiana 56 102条, N. langsdorffii× N. sanderae 12 448条, N. sylvestris 8583条, N. attenuata 355条, 其他烟草种453条。通过登陆GenBank, 将上述EST序列以FASTA格式下载保存, 作为烟草RGAs开发的原始数据。由于原始EST数据存在大量冗余序列, 为提高EST序列质量, 获得比原始EST序列更长, 且来自同一位点的共有序列, 采用TGICL工具拼接GenBank收录的烟草EST序列。结果由412 325条EST序列拼接出149 606条Uni-EST序列, 其中contigs 45 137条, singletons 101 169条, 序列最长为2312 bp, 最短为431 bp, 平均长度为874 bp。
利用tBlastn软件将112个已知的 R基因氨基酸序列与烟草Uni-EST序列比对分析表明, 比对E值小于或等于1E-10时有3个 R基因(RPW8.1, RPW8.2, xa27)无法检测到与之匹配的烟草Uni-EST, 其余109个 R基因共检测到6963条Uni-EST。由于具有相同结构域的 R基因序列存在相似性, 序列比对时具有相同结构域的 R基因常与同一Uni-EST序列匹配, 导致Uni-EST序列被重复计数。去除重复计数后, 共有1113个Uni-EST与109个 R基因序列匹配(数据未发表)。其中273个含NBS-LRR结构域, 546个含LRR-PK结构域, 53个含胞外LRR结构域, 102个只含PK结构域, 30个含Mlo结构域, 其余109个
| 表2 112个用于本研究的植物 R基因 Table 2 One hundred and twelve known Rgenes from plants used in this study |
| (续表2) 112个用于本研究的植物 R基因 Table 2 Continued One hundred and twelve known Rgenes from plants used in this study |
| (续表2) 112个用于本研究的植物 R基因 Table 2 Continued One hundred and twelve known Rgenes from plants used in this study |
未发现结构域。将与 R基因序列匹配的Uni-EST初步确定为烟草RGA, 并暂命名为NtRGA。
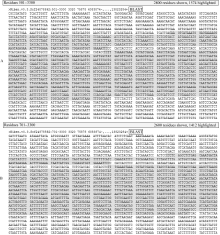
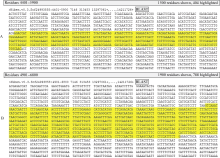
将NtRGA定位到烟草基因组上对分离特定候选抗病基因/QTL具有重要意义。随着 N. benthamiana全基因组草图的公布, 通过序列比对可直接将NtRGA定位到 N. benthamiana基因组上。用Blastn工具在 N. benthamiana全基因组草图中搜索与NtRGAs序列匹配的位点表明, 当分值大于50且E值小于1E-10时, 1113个NtRGA有1071个在 N. benthamiana基因组中找到相似的序列, 其中965个NtRGAs (90.7%)同时与基因组中多个片段匹配, 平均每个NtRGA与9.67个基因组片段匹配, 最多的一个(CL7158Contig2)与529个基因组片段匹配。鉴于植物基因在长期的进化过程中普遍存在多拷贝现象, 本研究取分值最高且E值最低的基因组序列作为NtRGA的基因组位点。最终1071个NtRGA被定位到712个基因组位点上, 共有218个基因组位点存在一个位点与多个NtRGAs配对(数据未发表)。进一步分析发现, 与同一个基因组位点配对的NtRGAs存在两种类型: (1)序列高度同源, 与基因组同一区段配对(图1); (2)序列无同源性, 与基因组同一位点不同区段配对(图2)。由于真核生物的外显子存在选择性剪切, 同一段基因序列可能会转录成不同的mRNA。属于类型1的NtRGA可能来自同一基因的不同转录本, 同时也有可能是不同烟草种间的同源基因。而类型2的NtRGA则可能来自同一基因的不同部位。EST通常只是完整基因的一部分, 因此, 来自相同基因的EST序列可能由于缺乏重叠区而无法被拼接在一起。
观察NtRGA在 N. benthamiana基因组中的位置表明, NtRGA在基因组中分布不均匀, 共有17处基因组区段存在多个NtRGA串联。例如全长63 kb的基因组scaffold (序列ID: Niben.v0.3.Scf25265845)包含4个NtRGA。
 | 图1 CL1421Contig1、CL1421Contig2与 N. benthamiana基因组序列比对结果A: CL1421Contig1与基因组scaffold (序列ID: Niben.v0.3.Scf24878592)的1078~1448 bp和1547~2749 bp区段匹配;B: CL1421Contig2与基因组支架Scf24878592的1276~1449 bp和1549~2786 bp区段匹配。Fig. 1 Alignment of CL1421Contig1 and CL1421Contig2 to N. benthamianagenomeA: CL1421Contig1 matches with 1078-1448 bp and 1547-2749 bp in genomic scaffold (sequence ID: Niben.v0.3.Scf24878592);B: CL1421Contig2 matches with 1276-1449 bp and 1549-2786 bp in genomic scaffold (sequence ID: Niben.v0.3.Scf24878592). |
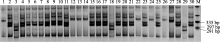
利用Perl脚本MISA从1113条NtRGA序列搜索SSR, 共发现78个SSR位点, 分布于72条序列, 其中含有SSR一个以上的NtRGA序列6条, 平均每 939 348 bp含1个SSR。根据SSR侧翼序列, 利用Primer Premier 5软件设计SSR引物, 共设计引物64对, 8条NtRGA序列由于SSR侧翼序列太短或者结构复杂而未能成功设计引物。新引物随后在24个烟草栽培品种和6个野生种中扩增, 并通过聚丙烯酰胺凝胶电泳检测其多态性(表3)。结果发现, 供试引物有54对在栽培种中能扩增出清晰条带, 其余10对无扩增产物或产生类似RAPD的非特异性条带。在可扩增引物中, 46对扩增产物长度与预期相近, 7对扩增产物长度大于预期, 另有1对扩增片段比预期短。可扩增的54对引物中有9对在烟草栽培品种间检测出多态性, 占可扩增引物的16.7%。检测等位基因数2~4个, 总共23个, 平均2.56个。54对在烟草栽培种中可扩增SSR引物在6个烟草野生种中均成功扩增。与栽培种相反, SSR引物在野生种中检测出较高的多态性, 共有41对引物检测出多态性, 占可扩增引物的75.9%, 检测等位基因数为2~4个, 总共92个, 平均2.61个。图3为引物RGA-63在烟草中的扩增带型, RGA-63在24个普通烟草资源和6个其他烟草种中均检测出4个等位基因。
 | 图2 FS423566和CL17188Contig1与烟草基因组序列比对结果A: FS423566与基因组scaffold (序列ID: Niben.v0.3.Scf24993858)的4904~5308 bp区段匹配; B: CL17188Contig1与基因组scaffold (序列ID: Niben.v0.3.Scf24993858)的5494~6282 bp区段匹配。Fig. 2 Alignment of FS423566 and CL17188Contig1 to N. benthamianagenomeA: FS423566 matches with 4904-5308 bp in genomic scaffold (sequence ID: Niben.v0.3.Scf24993858); B: CL17188Contig1 matches with 5494-6282 bp in genomic scaffold (sequence ID: Niben.v0.3.Scf24993858). |
作为“基因对基因”抗病机制的组成部分, 植物 R基因在识别病原物特异无毒基因编码产物中起重要作用[ 39]。本研究中从GenBank收录的烟草EST数据中成功鉴定出1113个RGA, 并通过序列比对将其定位到 N. benthamiana基因组上, 说明利用EST数据能够高效鉴定RGA。
过去几年, 通过数据挖掘已成功从甘蔗、小麦、玉米等作物鉴定RGA。Dilbirligi等[ 40]采用4种不同搜索策略在小麦中查找RGA序列, 包括结构域搜索, 单个或多个基序搜索, 共有序列搜索以及单一全长序列搜索。结果表明单一全长序列搜索效果最好。当E值≤ E-10, 采用单一全长序列共检测到243个含NBS-LRRs和101个其他类型的RGA序列。Xiao等[ 41]利用改良AFLP、RACE和数据挖掘方法鉴定玉米RGA和类 R基因EST时, 发现数据挖掘方法最为有效。Rossi等[ 42]通过严格的比对条件(E值
通过EST挖掘RGA的一个优点是所检测到的RGA均为表达的基因, 相反, 通过基因组序列挖掘的RGA存在不表达的假基因。Li等[ 32]从百脉根基因组鉴定的RGA中发现有65个编码不完整的蛋白序列, 最终被确定为假基因。Meyers等[ 10]从拟南芥基因组中搜索含NBS-LRR结构域的RGA, 发现至少有12个NBS-LRR基因由于框架移动和无义突变而演变为假基因。
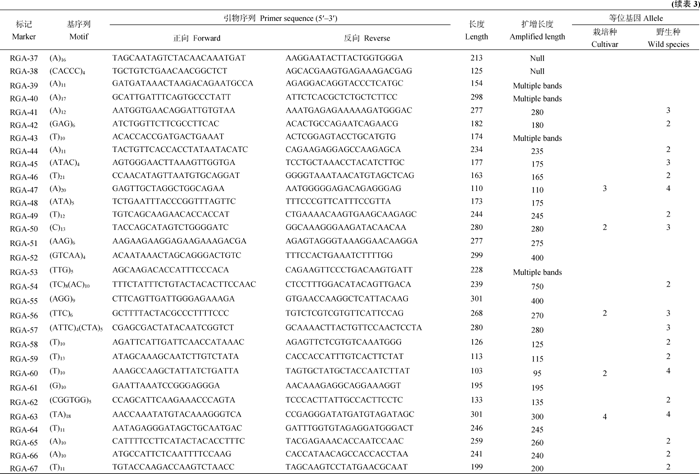
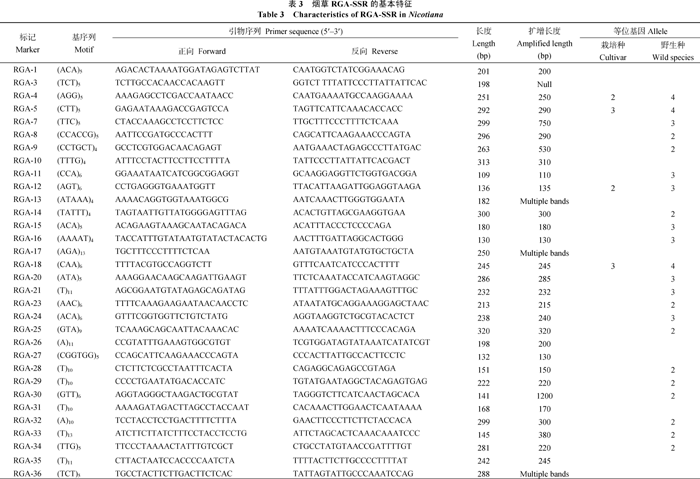 | 表3 烟草RGA-SSR的基本特征Table. 3 Characteristic of RGA-SSR in Nicotiana |
 | 图3 引物RGA-63在烟草中的扩增带型1: ATNARELLO; 2: KY26; 3: 大白筋599; 4: 白肋9号; 5: 阿波烟; 6: Margland 609; 7: 广黄5号; 8: 丰字1号; 9: 革新6号; 10: 革杂基; 11: 红花大金元; 12: 金星; 13: 白骨牛利; 14: 大秋根2; 15: 大山沟; 16:大同; 17: 大叶密合; 18: 大种白毛; 19: 2040; 20:74-16; 21: 夏湾娜; 22: Black Sea Samsun; 23: Burley Hampton; 24: Dark Virginia; 25: N. clevelandii A. Gray; 26: N. repandaWilld; 27: N. debneyi Domin; 28: N. rustica L.; 29: N. tomentosiformis; 30: N. sylvestris Speg. & Comes。Fig. 3 Amplification results of RGA-63 in Nicotiana1: ATNARELLO; 2: KY26; 3: Big White Burley 599; 4: White Burley 9; 5: Aboyan; 6: Margland 609; 7: Guanghuang 5; 8: Fengzi 1; 9: Gexin 6;10: Gezaji; 11: Honghuadajinyuan; 12: Jinxing; 13: Baiguniuli; 14: Daqiugen 2; 15: Dashangou; 16: Datong; 17: Dayemihe; 18: Dazhongbaimao; 19: 2040; 20: 74-16; 21: Xiawangna; 22: Black Sea Samsun; 23: Burley Hampton; 24: Dark Virginia; 25: N. clevelandii A. Gray; 26: N. repandaWilld; 27: N. debneyi Domin; 28: N. rustica L.; 29: N. tomentosiformis; 30: N. sylvestris Speg. & Comes. |
由于目前多数植物 R基因是通过图位克隆和转座子标签法克隆得到[ 43], 因此, 将RGA定位到基因组上显得十分重要。本文通过序列比对将其中的1071个NtRGA定位到 N. benthamiana基因组中712个位点上, 为后续烟草 R基因的克隆奠定了基础。本研究尚有42个NtRGA无法定位于 N. benthamiana基因组, 推测有以下原因: (1)目前公布的 N. benthamiana全基因组序列仅为草图, 尚有部分基因组区段未测序; (2)本研究鉴定的NtRGA仅部分来自 N. benthamiana, 来自其他烟草种的NtRGA由于种间基因组差异而无法定位。本研究通过分析NtRGA在基因组中的位置发现, RGA在基因组中的分布并不均匀, 部分RGA成簇存在。这与前人在其他植物中的研究结果相似[ 44, 45]。 R基因的串联方便 R基因的遗传变异, 有利于新 R基因的进化。Bertioli等[ 46]通过分析花生、百脉根和苜蓿发现反转座子与一些抗病基因簇关联。其他几个假设例如复制、基因转换和不等位交换已被用于解释 R基因成簇和进化[ 1]。
如何开发利用RGA0序列是RGA鉴定后需要解决的问题。本文通过SSR搜索从RGA序列中开发出62个RGA-SSR标记, 为后续抗病基因的定位克隆提供了有利的帮助。近期的研究表明, SSR在普通烟草具有较高的多态性, Bindler等[ 47]利用SSR构建普通烟草遗传图谱, 5119对SSR引物中有2415对(47%)在亲本间检测出多态性。本研究仅有16.7%的引物在普通烟草中检测到多态性, 且检测的等位基因数仅有2~4个。本研究开发的SSR标记多态性低的原因可能与标记来自表达基因序列有关。由于表达基因维持其生物学功能, 在进化过程中承受的选择压力远高于全基因的平均水平, 导致其序列高度保守。
利用植物 R基因蛋白质序列扫描EST数据是鉴定烟草表达RGA的有效途径。烟草表达RGA在基因组中分布不均匀, 其结构域以LRR-PK和NBS- LRR为主。烟草RGA-SSR在种内缺乏多态性, 但在种间具有较高的多态性。
| 1 |
|
| 2 |
|
| 3 |
|
| 4 |
|
| 5 |
|
| 6 |
|
| 7 |
|
| 8 |
|
| 9 |
|
| 10 |
|
| 11 |
|
| 12 |
|
| 13 |
|
| 14 |
|
| 15 |
|
| 16 |
|
| 17 |
|
| 18 |
|
| 19 |
|
| 20 |
|
| 21 |
|
| 22 |
|
| 23 |
|
| 24 |
|
| 25 |
|
| 26 |
|
| 27 |
|
| 28 |
|
| 29 |
|
| 30 |
|
| 31 |
|
| 32 |
|
| 33 |
|
| 34 |
|
| 35 |
|
| 36 |
|
| 37 |
|
| 38 |
|
| 39 |
|
| 40 |
|
| 41 |
|
| 42 |
|
| 43 |
|
| 44 |
|
| 45 |
|
| 46 |
|
| 47 |
|