


{kind=link}
{kind=link}
{kind=link}
利用基因组简约法开发烟草SNP标记及遗传作图
引用本文
肖炳光, 邱杰, 曹培健, 桂毅杰, 卢秀萍, 李永平, 樊龙江. 利用基因组简约法开发烟草SNP标记及遗传作图. 作物学报, 34(3): 397-404
XIAO Bing-Guang, QIU Jie, CAO Pei-Jian, GUI Yi-Jie, LU Xiu-Ping, LI Yong-Ping, FAN Long-Jiang. Development and Genetic Mapping of SNP Markers via Genome Complexity Reduction in Tobacco. 作物学报, 34(3): 397-404
Permissions
XIAO Bing-Guang, QIU Jie, CAO Pei-Jian, GUI Yi-Jie, LU Xiu-Ping, LI Yong-Ping, FAN Long-Jiang. Development and Genetic Mapping of SNP Markers via Genome Complexity Reduction in Tobacco. 作物学报, 34(3): 397-404
Copyright©2014, 作物学报编辑部
作物学报编辑部
利用基因组简约法开发烟草SNP标记及遗传作图
摘要
提出了一种基于基因组简约法开发SNP标记的方法, 即利用特定限制性内切酶酶切降低基因组复杂度, 利用高通量测序平台对酶切位点周围的目标片段进行富集测序, 设计一个生物信息学流程进行序列分析和SNP鉴定。以烤烟DH群体为例, 通过基因组简约法收集烟草基因组代表性片段和高通量测序产生11.4 Gb数据, 经生物信息学分析获得了1015个高质量SNP位点。以SSR标记为骨架, 绘制包括SNP标记在内、标记总数为1307的烤烟遗传连锁图。最后利用该遗传图谱和普通烟草2个祖先种的基因组序列, 分析烟草24个连锁群(染色体)之间的同源关系, 发现了大量染色体之间的重组或交换事件以及部分染色体之间的共线性。
关键词:
烤烟; SNP标记; 基因组简约法; 限制性内切酶; 遗传图谱
Development and Genetic Mapping of SNP Markers via Genome Complexity Reduction in Tobacco
Abstract
We proposed an approach for development of the SNP markers via genome complexity reduction in this study. The restriction enzymes were employed to digest target genome and then collect and sequence the fragments flanking the restriction sites by next-generation sequencing platform. A bioinformatics pipeline was developed for the SNP calling. A flue-cured tobacco DH population was used as a case to test the approach. The tobacco representative fragments were collected via a genome complexity reduction method and sequenced by using Illumina GA sequencer. A total of 1015 SNPs were found based on 11.4 Gb Illumina data using the bioinformatics pipeline. Taken available SSR markers (as backbone markers) together, a genetic linkage map with 1307 molecular markers was constructed. Large-scale inter-chromosomal (linkage group) DNA combinations or exchanges and several homologous pairs among the tobacco 24 chromosomes were detected based on the genetic map and the available genomic sequences of two tobacco (
Keyword:
Flue-cured tobacco; SNP marker; Genome complexity reduction; Restriction enzyme; Genetic map
普通烟草( Nicotiana tabacumL .)属于茄科( Solanaceae)烟草属( Nicotiana), 其基因组为异源四倍体, 是由2个野生种绒毛状烟草( N. tomentosiformis, T基因组)和林烟草( N. sylvestris, S基因组)杂交而来[ 1, 2], 基因组大小约为4.5 G[ 3], 其中含有大量的重复序列, 并且品种间遗传多态性非常低[ 4, 5]。由于烟草基因组的复杂性, 其分子标记开发进展较慢。迄今应用于烟草的分子标记主要包括RAPD[ 6]、AFLP[ 7]、SSR[ 8]、ISSR[ 9]、SRAP[ 10]、IMP[ 11]和DArT[ 12]等。这些标记已经成功应用于烟草种质遗传多样性分析、遗传图谱构建、重要性状基因定位/QTL分析等方面[ 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25]。但这些标记技术因自身存在的局限性限制了其进一步的发展与应用, 如通量小、精确性低、所需人力和时间成本高、在基因组上不能广泛分布等[ 26]。SNP (single nucleotide polymorphisms)是指在基因组上由单个核苷酸变异所引起的DNA序列多态性, SNP相较之前的标记不仅具有多态性好、能广泛均匀分布于全基因组的特点, 而且随着高通量测序及生物信息学数据分析技术的发展, 可以实现大规模自动化检测[ 27]。然而与先前的标记一样, SNP标记在对非模式物种的研究与应用中仍存在很大的困难和挑战[ 28]。近几年来, 逐渐出现了几种通过特异性酶切降低基因组复杂度、利用高通量测序技术对代表性文库测序的技术, 如RRL[ 29]、CRoPS[ 30]、RAD-Seq[ 31]和GBS[ 32]。这些技术不仅可以应用于有高质量参考基因组序列的模式物种上, 也可解决无参考基因组物种的标记开发及基因型分型难题[ 33]。
烟草分子标记遗传连锁图的构建始于21世纪初, Lin等[ 15]利用RFLP和RAPD构建了第一张烟草野生种间分子标记遗传连锁图。Nishi等[ 34]利用AFLP标记构建了第一张白肋烟分子标记遗传连锁图。肖炳光等[ 35]利用ISSR和RAPD标记构建了第一张烤烟遗传图谱。Bindler等[ 24]基于烤烟品种“Hicks Broad Leaf”和晒烟品种“Red Russian”间杂交产生的186个F2植株, 构建了一张由282个SSR标记组成、覆盖长度为1920 cM的遗传图谱; Bindler等[ 25]进一步开发了SSR标记, 并更新了之前的图谱。烟草包括烤烟、晒烟、香料烟、雪茄烟等不同类型, 烤烟是我国最主要的烟草类型(占烟草产量的80%以上)[ 36]。通过近年来的努力, 分别利用SSR标记和DArT标记, 获得了包含800余个分子标记的烤烟遗传连锁图[ 37, 22]。
本研究通过基因组简约法收集烟草基因组代表性片段和高通量测序, 设计了一个生物信息学分析流程鉴定SNP, 并利用获得的SNP数据对前期构建的烤烟SSR标记遗传图谱进行了加密和同源分析。
1 材料与方法
1.1 植物材料
本课题组前期建立的烤烟DH群体(包含207个DH株系), 其亲本为烤烟品种红花大金元(HD)和宽叶烟草(hicks broad leaf, HBL)[ 37], 本研究选取其中166个株系用于SNP标记开发分析。
1.2 基因组DNA提取与高通量测序
采用CTAB法提取烟草基因组DNA[ 38]。DNA利用限制性内切酶 Pst I和 Hpa II进行组合酶切, 收集长度500~1500 bp片段, 构建测序文库。采用Illumina GAII测序。
1.3 序列数据处理与SNP分析流程
1.3.1 测序数据的预处理 将测序获得的FASTQ格式文件首先利用Fastx软件进行质量过滤(Q>20)。接着切除用于区分样品4~8 bp的条形码序列, 考虑到Illumina产生的读序3°端错误率较高, 进一步切除了读序末端2~6 bp的碱基, 最终获得的读序长度为67 bp。为了便于后续序列联配, 保留了5°端5 bp的酶切位点序列。
1.3.2 SNP检测及基因型分型 利用Usearch软件[ 39]对亲本(HD和HBL)及DH群体各株系的读序进行序列联配(聚类), 允许的错配数为1 bp。为了保证后续分析的质量, 选取读序覆盖度在3~50的联配结果(簇)用于生成株系的一致性序列。在获取个体样品序列聚类结果的一致性序列时采用了如下方法: 将每个簇中的序列用Usearch进行联配, 若某个碱基位点错配数目≥3或≥25%, 即用此位点上所含碱基的兼并码(如W代表A和T)表示; 若错配数目未达到该标准, 则该碱基位点用最多出现的碱基表示。
将所获得的株系一致性序列汇总, 利用Usearch软件进行序列联配, 一致性序列之间允许1个碱基的错配。通过筛选获得亲本至少存在一个、并且含有至少30个DH株系一致性序列支持的簇用于SNP检测及基因型分型。对于每一个候选簇, 其中若某一碱基位点出现的普通错配碱基个数≥3, 则将该碱基位点视作简单SNP (simple SNP); 若某一碱基位点出现简并码(WRSKMY)个数≥3, 将该碱基位点视作杂合SNP (hemi-SNP)[ 40](图1)。然后把在一个簇中变异个数超过2次的结果筛除。通过自制Perl脚本实现基因型分型。
 | 图1 烟草基因组中两种SNP (简单和杂合SNP)模式图图中S和T分别代表烟草2个祖先基因组(以本研究由HD和HBL构建的DH群体为例)。Fig. 1 Two types of SNPs (simple and hemi-SNP) in tobacco genomeS and T represent genomes of two tobacco progenitors, respectively (tobacco DH population from offspring of HD × HBL). |
1.4 遗传图谱构建及同源分析
将获得的SNP基因型分型结果进行PIC (polymorphism information content)值检验, 筛选PIC> 0.375的标记用于构建遗传连锁图, 同时以Tong等[ 37]构建的遗传连锁图上611个SSR标记作为骨架标记, 利用JoinMap 4.0[ 41]构建遗传连锁图, 采用回归作图算法计算标记的顺序及遗传距离, 参数设定为Kosambi函数, LOD值≥7, 并利用MapChart2.2[ 42]绘制遗传图谱。
由于Bindler等[ 25]开发的SSR标记是基于美国烟草基因组测序项目TGI (Tobacco Genome Initiative)数据库(http://www.pngg.org/tgi/)中的序列, 为了获取其SSR标记全长序列以便于后续分析, 将其SSR标记左、右引物分别锚定至TGI序列上, 分别找出左、右引物序列对应TGI序列的5个最佳匹配序列(-p BlastN-m8-b5-v5), 根据其匹配序列的位置及正反向信息, 提取在同一拼接序列且左右引物序列之间序列长度大于50的序列。由于Tong等[ 37]开发的标记除了基于TGI序列外还根据EST序列设计, 故下载了相应序列数据, 并用同样方法获取其SSR标记全长序列。将构建的遗传图谱上的分子标记序列和Bindler等[ 25]构建的标记序列分别锚定到(-p BlastN-m8-b1-v1-e1e-20)已拼接完成的2个烟草祖先种的基因组序列(国家烟草基因研究中心)上进行染色体进化及同源性分析。
2 结果与分析
2.1 DH群体测序结果及其SNP检测分析
利用Illumina GAII共获得11.4 Gb原始测序数据。对原始读序进行质量过滤及序列预处理后, 对
各个株系的读序进行序列联配。汇总联配结果后, 统计各联配的读序数目, 发现56.1%的联配中只有1~2条序列, 而包含3~50条读序的联配占39.7%, 说明在本实验中通过酶切测序的位点中, 有约40%位点测序达到了3~50倍的覆盖。为了保证后续分析的质量, 并减少重复序列的影响, 仅将这些包含3~50倍覆盖度的联配序列(簇)用于下步分析。
在得到每个株系一致性序列后, 统计每个株系一致性序列的个数, 获得亲本及80个DH株系一致性序列的平均个数为21 039个, 即平均每个株系基因组上有2万多个酶切位点的测序覆盖度达到3~50倍覆盖。
将80个DH株系和2个亲本的一致性序列汇总、联配并筛选2个亲本至少存在一个的联配结果(图2), 发现共有42 347个符合要求的联配结果, 其中某一位点仅含1~5个株系的一致性序列占多数(62%)。为保证株系个数, 选取至少含30个株系一致性序列的联配结果, 最终获得10 623条候选联配结果用于SNP检测及基因型分型。进一步分析发现, 867个序列联配中变异数目为1~2个, 经过统计发现变异碱基位点总数为1015个, 即大致可以推断在烟草基因组上每701个碱基(10 263×67/1015)出现一个SNP变异; 这1015个SNP中, 158个为简单SNP位点, 857个为杂合SNP位点。
2.2 利用SNP标记构建烤烟遗传图谱
以Tong等[ 37]构建的含611个SSR标记的遗传连锁图为骨架, 加入1015个SNP候选标记并整合Lu等[ 22]构建的315个DArT标记, 最终获得一张包含1307个分子标记的遗传图谱(图3, 限于篇幅仅列出部分连锁群)。
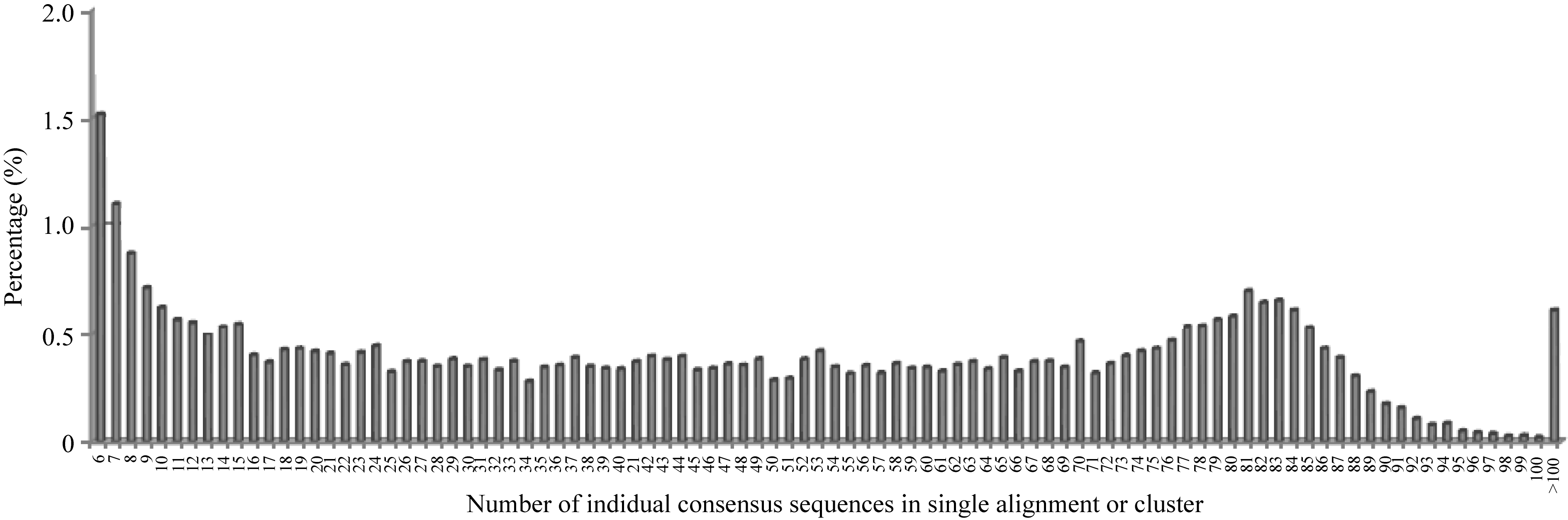 | 图2 含有不同株系一致性序列个数的联配比例分布Fig. 2 Percentage of alignments or clusters with different numbers of consensus sequences of individuals |
 | 图3 基于SSR、DArT和SNP标记的烤烟遗传连锁图限于篇幅仅列出5个连锁群, 数字“10”开头表示SNP标记; TM、PT开头表示SSR标记; DT开头表示DArT标记; 后缀“_”表示与Bindler等[ 25]遗传图谱共有的标记。Fig. 3 Genetic linkage map of flue-cured tobacco based on SSR, DarT, and SNP markersFive linkage groups are selected because of limited pages. The markers with “10”, “PT/TM”, “DT” refer to SNP, SSR, and DArT markers, respectively. Markers with “_” as suffix are ones sharing with genetic map by Bindler et al.[ 25] |
该遗传图谱总长3002 cM, 标记密度为2.3 cM, 其中SNP标记555个(表1)。总体上, SNP标记均匀分别在各个连锁群上(平均每个连锁群23.1个, 其中有3个连锁群上的SNP数量超过40个, 另有3个连锁群上的SNP数量少于10个)。在166个株系中, 所有SNP标记的平均基因型缺失率为7.1%。SNP标记的加入, 基本没有改变原有SSR标记遗传连锁图的骨架[ 37], 验证了本研究开发的烟草SNP标记的可靠性和可用性。
2.3 同源分析
将本研究中构建的遗传图谱和Bindler等[ 25]构
建的遗传图谱标记分别在普通烟草祖先种基因组(T和S基因组)拼接序列上定位的结果可以分为2种情况: 一是部分标记可以在S和T基因组同时定位; 二是部分标记只能在其中一个基因组中定位, 仅能在一个祖先基因组上定位的标记数量列于表2。由表2可知: (1) 2张图谱标记在2个祖先基因组上定位的数量分布趋势基本一致。(2)遗传图谱标记定位在2个祖先基因组上的数量虽有差别, 但在2个祖先种基因组上均有分布。该结果表明, 自2个祖先种杂交形成四倍体烟草以来, 来自2个祖先种的24条染色体之间DNA的交换或染色体重组非常频繁, 远不
| 表1 利用SNP等标记构建的烤烟遗传图谱 Table 1 Linkage map of flue-cured tobacco with SNP and other molecular markers |
止Bindler等[ 25]建议的4次重组。(3)第22染色体(连锁群)等几个染色体明显发生了S/T基因组间的大片段重组。
在进行标记定位祖先种基因组过程中, 发现有些来自不同连锁群的分子标记可以定位在同一条祖先基因组拼接序列上。这一定位结果可能是这2个染色体具有高度同源性导致的序列相似性造成的。为此统计了2个连锁群标记定位在同一祖先种基因组序列的数量(表3)。很明显, 有些染色体间表现出明显的共线性或同源性。基于该结果, 推测出部分染色体间同源性(共线性)的大致关系, 如连锁群3和17, 7和19等。其中一些染色体与多个染色体具有共线性, 可能由染色体重组造成。另外, 有些连锁群(如第24连锁群)可能因丢失而没有明显的同源染色体。
3 讨论
本研究提出了一种基于基因组简约法开发SNP标记的方法, 并在烟草上开发了SNP标记, 与其他标记整合获得了包含1307个标记的高密度烤烟遗传连锁图。利用该遗传图谱和Bindler等[ 25]构建的遗传图谱标记比对2个烟草祖先种序列, 采用生物信息学方法发现了目前的栽培烟草中24条染色体与祖先种染色体之间的对应关系, 同时也找出了这24条染色体之间存在的共线性或同源关系, 为今后深入研究栽培烟草的起源进化奠定了基础。
本研究开发的SNP标记是基于高通量测序平台的基因组简约技术。与另外几种通过测序获得SNP基因型的技术一样, 主要在于选择特异性限制性内切酶或组合对整个基因组进行酶切, 利用酶切位点
| 表2 普通烟草遗传图谱标记仅能在一个基因组(S和T基因组)特异定位的标记数量 Table 2 Number of linkage map markers which could only be mapped in one of two ancient genomes (S/T genome) |
| 表3 来自2个连锁群标记定位在同一条祖先种基因组拼接序列的数量 Table 3 Scaffold number anchored by markers from two linkage groups |
附近的序列构建代表性文库, 进而实现对整个基因组上的特定位点序列的富集和测序。对于植物而言, 可选择甲基化敏感的酶, 从而避免酶切基因组上的重复区域[ 33]。对于基因组已测序的物种, 可以根据参考基因组和所选限制性内切酶序列估测酶切位点个数, 从而设定测序量大小。然而对于无参考基因组的物种, 无法精确获得酶切位点个数, 因而无法估计测序量的大小, 有待开发新的方法。本研究在前期开发烟草DArT标记的基础上[ 22], 摸索获得了烟草基因组简约的最佳酶切组合。由于不同植物基因组序列构成不同, 需要不同的酶切方式以求达到最佳代表性基因组序列样本。在本研究的测试流程中, 发现在个体样品序列联配结果中, 有近55%的联配因只含有1~2条序列而被过滤。若对酶切位点个数能有较好的预估, 适当增加测序量, 提高测序深度, 将会大大提高候选位点的个数, 相应改善SNP检测以及基因型分型效率。此外, 还需进一步优化SNP检测方法, 尤其是对1~3个碱基插入缺失的鉴定。这种变异也广泛存在于基因组中, 据估计其比例约为SNP数目的50%, 若能加以利用将大大增加总标记数目。
4 结论
利用基因组简约法开发与分析烤烟DH群体的SNP标记, 以SSR标记遗传连锁图为骨架, 获得包括SNP标记在内、标记总数为1307的烤烟遗传连锁图。利用该遗传图谱和普通烟草两个祖先种的基因组序列分析24个连锁群(染色体)之间的同源关系, 发现了大量染色体之间的重组或交换事件以及部分染色体之间的共线性。
致谢: 感谢Diversity Arrays Technology Pty Ltd公司Adrzej Kilian博士对本研究的技术支持。
The authors have declared that no competing interests exist.
作者已声明无竞争性利益关系。The authors have declared that no competing interests exist.
参考文献
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|