

 Aij/loci) for A, B and D genomes were 6.88, 7.92 and 7.62, respectively. Their average genetic dispersion indices (Ht) were 0.637, 0.694 and 0.656, respectively. The B genome showed the highest genetic diversity among the three wheat genomes. The landraces had a higher genetic diversity than the modern varieties, and the major difference between the landraces and the modern varieties in China existed in the D genome, followed by B and A genomes. The majority of the accessions (65.6%) had heterogeneity at the 112 loci detected. The highest heterogeneity locus percentages were 9.09 and 12.73 in the modern varieties and the landraces, respectively. SSR data were analyzed with NTSYS-pc software. The genetic similarities between accessions were estimated with the DICE coefficient. The accessions clustered into two groups, the modern varieties and the landraces by the un-weighted pair-group method using arithmetic average (UPGMA). The trend of correlation coefficients between genetic similarity matrices based on different numbers of random alleles and that of 802 alleles showed that 550 alleles were sufficient to construct a robust dendrogram. The separated simulations from six sub-samples revealed that 550 alleles were the minimum number required to confidently determine the genetic relationships. It was shown that the number of alleles (loci) needed do not have a strong association with the number of wheat lines in the sample size. These data suggested that 73 loci with good polymorphism are needed to reflect genetic relationships among accessions with more than 90% certainty. In the dendrogram, most accessions from the same wheat region were clustered together, and those from geographically adjacent regions usually appeared in the same small group. This indicated that genetic diversity of Chinese common wheat has a close association with their geographic distribution and ecological environment.
Aij/loci) for A, B and D genomes were 6.88, 7.92 and 7.62, respectively. Their average genetic dispersion indices (Ht) were 0.637, 0.694 and 0.656, respectively. The B genome showed the highest genetic diversity among the three wheat genomes. The landraces had a higher genetic diversity than the modern varieties, and the major difference between the landraces and the modern varieties in China existed in the D genome, followed by B and A genomes. The majority of the accessions (65.6%) had heterogeneity at the 112 loci detected. The highest heterogeneity locus percentages were 9.09 and 12.73 in the modern varieties and the landraces, respectively. SSR data were analyzed with NTSYS-pc software. The genetic similarities between accessions were estimated with the DICE coefficient. The accessions clustered into two groups, the modern varieties and the landraces by the un-weighted pair-group method using arithmetic average (UPGMA). The trend of correlation coefficients between genetic similarity matrices based on different numbers of random alleles and that of 802 alleles showed that 550 alleles were sufficient to construct a robust dendrogram. The separated simulations from six sub-samples revealed that 550 alleles were the minimum number required to confidently determine the genetic relationships. It was shown that the number of alleles (loci) needed do not have a strong association with the number of wheat lines in the sample size. These data suggested that 73 loci with good polymorphism are needed to reflect genetic relationships among accessions with more than 90% certainty. In the dendrogram, most accessions from the same wheat region were clustered together, and those from geographically adjacent regions usually appeared in the same small group. This indicated that genetic diversity of Chinese common wheat has a close association with their geographic distribution and ecological environment. Synthetic
Synthetic ×
× {kind=link}
{kind=link}
宁麦9号对其衍生品种的遗传贡献
[姜朋 , 陈小霖, 张平平, 张鹏, 姚金保, 马鸿翔
, 陈小霖, 张平平, 张鹏, 姚金保, 马鸿翔* ]
, 陈小霖, 张平平, 张鹏, 姚金保, 马鸿翔]
|
|


*通讯作者(Corresponding author): 马鸿翔, E-mail:hxma@jaas.ac.cn
第一作者联系方式: E-mail:hmjp2005@163.com, Tel: 025-84391793
宁麦9号是江苏省农业科学院选育的优质弱筋专用小麦品种, 具有高产、稳产和广泛的适应性及抗小麦黄花叶病、赤霉病等特点, 近年来已成为江苏淮南麦区小麦育种的重要亲本。为了解宁麦9号对新育成小麦品种的遗传贡献, 利用170对SSR引物, 对宁麦9号及其9个衍生品种进行全基因组扫描分析。共检测到471个等位变异位点, 每对引物可检测1~6个, 平均2.8个。UPGMA聚类分析表明, 扬麦18与宁麦9号遗传距离最小, 扬辐麦4与宁麦9号遗传距离最大。遗传相似系数表明, 在人工选择的作用下, 这些以宁麦9号为亲本育成品种的遗传背景一半以上来自宁麦9号。宁麦9号等位变异在其衍生材料中的分布比例因品种而异, 且不同染色体间差异较大, 但相同位点的等位变异比例在A、B、D染色体组间相近。全基因组SSR扫描分析发现, 10个标记位点在宁麦9号和由其选育的9个新品种具有完全相同的扩增带型, 其中9个位点与重要农艺性状相关, 或与控制优良性状的基因紧密连锁。将宁麦9号与主要弱筋小麦生产品种宁麦13 (宁麦9号系选)进行基因组比较, 两者遗传相似系数达0.732, 说明宁麦13与宁麦9号遗传背景高度相似, 而且两品种在蛋白质含量和抗赤霉病位点相关标记位点高度一致, 这一结果可以部分解释宁麦13同样具有的抗赤霉病与优质弱筋的优点。
Ningmai 9 is a soft wheat variety with high yield, wide adaption, and resistance to multiple diseases including
宁麦9号是江苏省农业科学院以扬麦6号为母本、日本小麦品种“西风”为父本杂交, 采用集团选择法选育而成, 因具有高产、稳产和广泛的适应性、优质弱筋专用品质及抗小麦黄花叶病、赤霉病等特点, 已成为我国江苏省的主栽品种[ 1]。宁麦9号也是我国小麦育种的重要亲本。以该品种为亲本育成了一批适于长江中下游麦区种植的小麦新品种, 自2006年以来通过国家或省审定的品种已达12个, 如宁麦13、宁麦14、宁麦16、生选4号、生选6号、扬麦18、扬麦21、扬辐麦4号、南农0686、宁麦18、镇麦5号、镇麦8号等, 其中宁麦13是近年江苏省种植面积最大的弱筋品种。2010—2012年江苏省淮南组区域试验36个品种中带有宁麦9号遗传背景的材料达18个, 显示出宁麦9号作为新一代骨干亲本的潜力, 及对选育高产、优质、抗病小麦新品种的价值, 因此, 研究其基因组组成对衍生品种的遗传贡献对该亲本的育种应用和新品种选育具有重要意义。
分子标记技术的发展为从全基因组层次分析亲本对衍生品种的遗传贡献提供了重要工具。特别是SSR (简单重复序列)标记, 由于其具有数量丰富、多等位基因、共显性遗传、全基因组分布等特点, 被广泛应用于作物遗传多样性、核心种质构建、关联
分析、骨干亲本等研究[ 2, 3, 4, 5, 6, 7]。目前, 在普通小麦基因组中已开发了大量SSR标记, 并利用这些标记对小麦骨干亲本及其衍生后代品种(系)进行了遗传分析, 如燕大l817[ 8]、鲁麦14[ 9]、洛夫林10号[ 10]、阿夫[ 11]、碧蚂4号[ 12], 发现骨干亲本普遍含有一些遗传性较强的基因组区段, 其衍生系的遗传多样性随世代的增加而降低, 通过分析小麦骨干亲本的遗传特点及对衍生品种的遗传贡献, 为在育种中进一步有效利用这些骨干亲本提供了依据。宁麦9号作为近年来江苏淮南麦区的重要亲本, 其遗传物质在育种世代中的传递特点及对衍生品种的遗传贡献至今未见报道。本研究采用覆盖小麦全基因组的SSR标记, 对宁麦9号及其衍生品种进行遗传背景扫描, 探讨宁麦9号基因组片段在其衍生品种中的分布, 尤其是对宁麦13等主推品种的遗传贡献, 从而为亲本选配和新品种培育提供依据。
以宁麦9号及其9个衍生品种为试验材料(表1)。选取每个品种幼嫩的叶片5~6 cm, 采用SDS-酚法[ 13]提取DNA。
| 表1 供试材料名称、来源及审(认)定时间 Table 1 Name, pedigree, and released year of varieties tested |
共筛选到覆盖全基因组的170对多态性SSR引物, 每条染色体3~14对, 平均8对。PCR反应体系为10 μL, 包含10×buffer 1 μL、15 mmol L-1 MgCl2 0.6 μL、2 mmol L-1 dNTP 0.8 μL、20 ng μL-1引物1 μL、0.1 μL Taq酶、20 ng μL-1模板DNA 3 μL、去离子水3.5 μL。PCR扩增程序为94℃预变性5 min; 然后94℃变性45 s, 50~60℃ (依引物而定)退火45 s, 72℃延伸90 s, 36个循环; 最后72℃延伸10 min。扩增产物经聚丙烯酰胺凝胶电泳检测。在相同位点上, 有带记为1, 无带记为0, 缺失记为9。
利用NTSYSpc version 2.10t软件计算遗传相似性系数, 用UPGMA法进行聚类分析。
依据Somers等[ 14]的遗传连锁图估计SSR位点间的遗传距离, 且每条染色体的长度与该遗传连锁图一致, 并在图中标注本研究未覆盖的区间。采用Microsoft Excel VBA (MapDraw V2.1)绘制遗传连锁图。
UPGMA聚类分析显示, 宁麦9号与扬麦18遗传距离最小, 其次与宁麦18, 再次与宁麦16, 与扬辐麦4号遗传距离最大(图1)。遗传相似系数与聚类结果一致(表2), 例如, 与宁麦9号的遗传相似系数, 扬麦18最高(0.857), 扬辐麦4号最小(0.539), 说明这些以宁麦9号为亲本新育成的品种的遗传背景一半
以上来自宁麦9号。在两两比较中, 宁麦13与生选6号遗传相似系数最高, 二者也最先聚为一类(图1)。
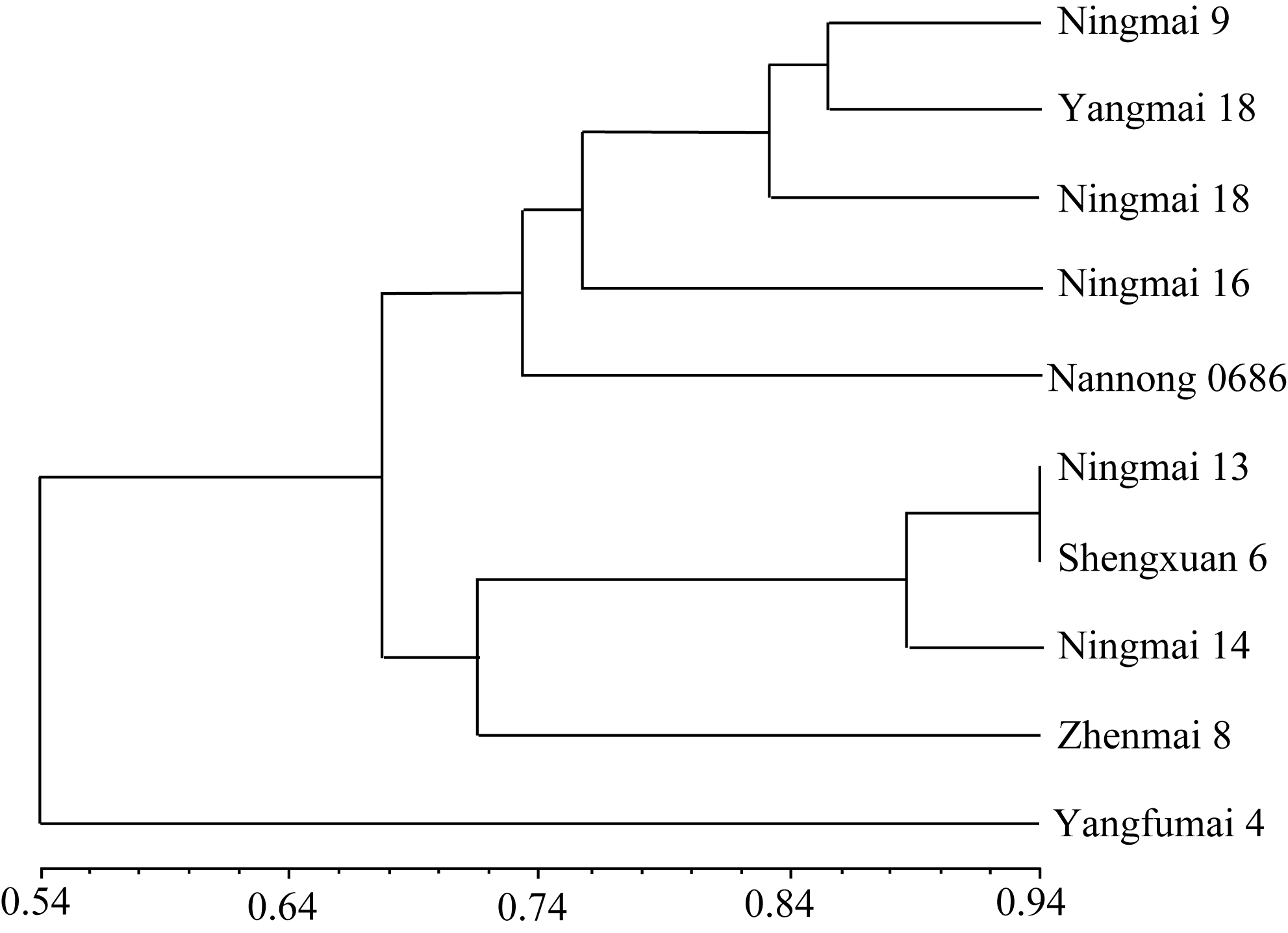 | 图1 宁麦9号与衍生品种的UPGMA聚类分析Fig. 1 UPGMA cluster analysis of Ningmai 9 and derived varieties |
| 表2 宁麦9号与其衍生品种间的遗传相似系数 Table 2 Genetic similarity coefficient among Ningmai 9 and derived varieties |
170对SSR引物共扩增出471个等位变异位点, 每对引物1~6个等位变异, 平均2.8个。不同品种及不同染色体间存在较大差异(表3)。1A染色体上, 南农0686与宁麦9号相同等位位点比例达85%, 而镇麦8号与宁麦9号该比例仅23%; 不同染色体上宁麦9号与新育成品种间的相同等位变异位点平均比例为56%~86%, 超过80%的有3条染色体, 超过70%的有12条染色体, 仅有2条染色体不超过60%。在扬麦18和南农0686中, 各有3条染色体的相同等位变异位点比例达到100%。A、B、D染色体组相同等位变异位点平均比例相近, 分别为70%和73%、71%。全基因组SSR扫描分析发现, 宁麦9号中有10个标记位点与其9个衍生品种的带型完全相同, 分布于1A、2A、2B、2D、3A、4A、4D、7A和7B染色体上(图2)。
宁麦13是以宁麦9号经系统选育而成, 其与宁麦9号遗传相似系数达0.732 (表2), 各染色体上相同等位变异比例为54%~95% (表3), 仅1A、1D和3D染色体的相同等位变异比例低于60%。A、B、D基因组的相同等位变异比例十分接近, 分别为73%、73%和72%。宁麦13的多个染色体区段保留了宁麦9号的遗传信息, 特别是1B、2D、4A、4B染色体的大部分区段与宁麦9号相同(图2)。
| 表3 不同染色体上宁麦9号与衍生品种间的相同等位变异比例 Table 3 Percentage of alleles shared between Ningmai 9 and its derived varieties on 21 linkage groups (%) |
骨干亲本的确定都源自育种家对育成优良品种系谱的总结, 对指导育种工作的亲本选配有重要意义, 骨干亲本之所以能选出众多优良品种, 除其本身具有优良性状基础外, 其重要农艺性状的基因组片段必须具有较强的遗传传递能力, 从经典的遗传学角度分析, 这些性状的遗传需要具有较高的一般配合力[ 15]。目前, 通过配制多个杂交组合, 已对宁麦9号的产量、品质和抗病性遗传机制进行了初步研究。例如, 利用宁麦9号、扬麦158等8个小麦亲本材料进行双列杂交, 宁麦9号的不孕小穗数(负向效应)主穗粒数、小穗粒数、每穗粒数、单穗粒重和收获指数等6个性状的一般配合力均居8个亲本之首, 且具有较多控制穗粒数和收获指数遗传
的显性基因, 说明宁麦9号对产量有利的基因位点多, 加性效应大[ 16]; 利用宁麦9号等7个小麦品种配制双列杂交, 宁麦9号在不同遗传背景下, 表现穗粒数的一般配合力最好, 且具有控制穗粒数遗传较多的显性基因[ 17]; 以江苏省育成的7个弱筋小麦品种与硬度、蛋白质含量、面筋强度、碱水保持力各不相同的6个常用优质亲本杂交, 研究了蛋白质含量和碱水保持力的遗传, 结果宁麦9号的蛋白质含量和碱水保持力的一般配合力最好, 能够显著降低杂种后代的蛋白质含量和吸水率, 且具有控制蛋白质含量和碱水保持力遗传最多的显性基因[ 18, 19]; 对小麦赤霉病[ 20]和梭条花叶病的抗性[ 1], 宁麦9号都具有较高的一般配合力。本研究利用基因型数据对以宁麦9号为亲本育成的品种分析, 除杂交种子辐射诱变产生的品种外, 通过杂交育成的品种与宁麦9号的遗传相似性较高, 为0.619~0.857, 均高于60%, 说明宁麦9号的遗传成分较多地保留在衍生品种中。
 | 图2 宁麦9号及其衍生品种的遗传连锁图灰色区域为宁麦13和宁麦9号相同的基因组区段, ◆表示宁麦9号与其衍生品种的相同标记位点。Fig. 2 Genetic linkage map of Ningmai 9 and its derivatesGray regions are identical linkage blocks between Ningmai 13 and Ningmai 9. ◆ shows the identical loci between Ningbai 9 and its derivates. |
SSR分子标记将以宁麦9号为亲本育成的品种分为三大类。理论上讲, 遗传相似系数与品种聚类主要取决于系谱[ 21]。扬麦18与宁麦9号相似系数最好, 主要因为其在最后一轮杂交程序中与宁麦9号进行了4次回交, 使后代中宁麦9号的遗传背景大幅上升。具有相同系谱来源的品种本应聚为一类, 但本研究中同一系谱来源的品种并非如此, 如宁麦16与生选6号均为宁麦8号/宁麦9号杂交后代, 但二者的遗传相似系数仅0.651, 育种选择是一个重要原因。宁麦8号为大穗矮秆型, 宁麦9号为多穗丰产型品种。宁麦16在继承宁麦9号多穗的基础上改良了熟相; 生选6号则提高了抗倒性和赤霉病抗性, 宁麦9号携有赤霉病抗性基因 Fhb1[ 22], 而宁麦8号中则含有较多控制赤霉病抗性的隐性基因[ 20]。生选6号赤霉病抗性的提高可能由于其既含有来自宁麦9号的 Fhb1, 又聚合了宁麦8号的其他抗病位点。宁麦13和宁麦14为宁麦9号系选材料, 其与宁麦9号的遗传相似系数低于扬麦18、宁麦18等品种, 可能由于其变异来源于天然异交[ 23]。这一组合的杂交种子经60Co-γ辐射诱变, 从后代中选出扬辐麦4号; 辐射诱变产生了新的遗传变异, 因此扬辐麦4号又归为另一类群, 在所有供试品种中与宁麦9号遗传相似系数最低, 说明辐射诱变是创造新的遗传变异的有效手段[ 24]。
SSR标记结果显示, 在10个标记位点上, 宁麦9号与其9个衍生品种完全相同。参考已报道的小麦重要性状QTL连锁图谱, 除cfa2037外, 其余标记位点都与重要性状相关, 或是与控制某些性状的基因紧密连锁。在1A染色体上, wmc24标记关联到株高、穗型等[ 25]; 2A上的wmc819与千粒重相关联[ 26]; 2B上的wmc296标记与根干重QTL连锁[ 27], wmc317与控制株高的基因 Rht4连锁[ 28]; 在2D的wmc111标记相邻区段检测到控制籽粒蛋白含量和吸水率的QTL[ 29]; 4A上wmc313标记与抗叶锈病基因 Lr28紧密连锁[ 30]; 4D上的wmc473标记与抗赤霉病QTL关联[ 31]; 7A上的wmc83可作为选择小麦不孕小穗数的分子标记[ 32]; 7B上的wmc526与控制籽粒色素含量[ 33]和抗穗发芽[ 34]的基因相关联。宁麦9号在这些标记上是否存在优良等位变异位点, 需通过关联分析才能得出可靠的结论, 但结合前人的研究可以推测这些标记位点所在的染色体区域可能为与小麦重要农艺性状相关的QTL热点区域。
作为江苏淮南麦区的主要小麦品种之一, 宁麦13曾在2008年和2011年两次创造淮南地区红麦高产记录(8430 kg hm-2和9315 kg hm-2), 是目前江苏淮南麦区面积最大的弱筋小麦品种。Lin等[ 35]在2A的wmc181、7A的wmc83和7B的wmc476相邻区段检测到抗赤霉病QTL, 本研究发现宁麦13与宁麦9号在这几个标记位点有相同的等位变异; 此外, 在2B的gwm257[ 36]和4A的wmc397[ 37]标记相邻区段报道了与籽粒蛋白含量有关的QTL, 宁麦13与宁麦9号在这个标记位点表现也无差异。这一结果或许可以部分解释宁麦13遗传了宁麦9号的抗赤霉病性和优质弱筋性状。对纹枯病的抗性, 宁麦13与宁麦
9号则有较大差异, 本研究发现两品种在标记位点gwm77 (3A)和gwm508 (6B)上有不同扩增结果, 而在这2个标记附近检测到抗纹枯病QTL[ 38], 可能是两品种有不同的纹枯病抗性原因之一。
在9个以宁麦9号为亲本育成的小麦新品种中, 宁麦9号的遗传成分占一半以上, 遗传贡献很大。有10个标记位点是宁麦9号和衍生品种共有的位点, 而这些位点几乎都与曾报道小麦重要性状QTL相关, 可能是重要性状的热点区域, 宁麦9号在这些位点可能具有优异等位变异。宁麦13遗传了很多宁麦9号的优良等位变异, 值得深入研究, 有望在今后淮南麦区新品种选育中发挥作用。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|