


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
普通菜豆基因组SSR标记开发及在豇豆和小豆中的通用性分析
[陈明丽1  , 王兰芬
, 王兰芬1 , 武晶1 , 张晓艳2 , 杨广东3 , 王述民1, * ]
, 王兰芬]
|
|


*通讯作者(Corresponding author): 王述民, E-mail:wangshumin@caas.cn, Tel: 010-62175628
第一作者联系方式: E-mail:sscmlss_cool@126.com
分子标记具种属间通用性可提高其利用效率, 并降低标记开发成本。本研究基于Roche 454超高通量测序技术获得普通菜豆基因组测序结果, 共开发了560个普通菜豆基因组SSR标记。利用2份普通菜豆品种对标记进行初步筛选, 有421个标记能够有效扩增。用新开发的标记分析16份豇豆和16份小豆的通用性。结果显示, 185个普通菜豆基因组SSR标记在豇豆中能有效扩增, 通用性比率为43.9%; 161个SSR标记在小豆中能有效扩增, 通用性比率为38.2%; 在豇豆和小豆中都能获得有效扩增条带的标记共138个; 并且普通菜豆基因序列SSR标记在豇豆和小豆中的通用性比率高于基因间序列SSR标记。通用性标记的多态性分析表明, 豇豆和小豆的多态性比率分别为34.0%和24.8%; 且豇豆和小豆中基因间标记的多态性都比基因内标记的多态性高。上述通用性标记为豇豆属作物的多样性评价、连锁图谱的构建及基因定位等方面的研究提供了便利。
Transferability analysis of molecular markers has significantly improved their development efficiency and reduced their development cost. A total of 560 novel SSR markers were successfully developed based on common bean genomic sequences, and 421 (75.2%) of those markers generated effective amplification bands from two accessions of the cultivated common bean. Two
普通菜豆、豇豆及小豆均是我国的主要栽培食用豆种。普通菜豆属于豆科(Leguminosae), 蝶形花亚科(Papilioni-deae), 菜豆族(Phaseoleae)菜豆属( Phaseolus)[ 1]。豇豆[ 2]和小豆[ 3]属豆科(Leguminosae), 蝶形花亚科(Papilionideae), 菜豆族(Phaseoleae), 豇豆属( VignaL.)。豇豆属含有2个亚属, 豇豆属于 Vigna亚属, 小豆属于 Ceratoropis亚属。
豇豆和小豆在中国栽培历史悠久, 全国各地都有种植。中国被认为是豇豆的起源中心或次级起源中心之一, 目前除西藏自治区外, 其他各地都有豇豆生产[ 4]; 小豆原产中国, 在中国华北、东北、黄河及长江中下游地区广泛种植[ 5]。豇豆和小豆在我国农产品出口, 饮食改善以及作物种植结构优化等方面具有重要作用[ 6]。但是, 与其他豆科模式作物如普通菜豆和苜蓿相比, 豇豆[ 7]和小豆[ 8]公开发表的分子标记数目极少。
分子标记在许多作物的遗传研究中起重要作用。SSR标记因其在基因组中分布广泛且具有共显性遗传、可重复性、多态性丰富等特点而被认为是最好的分子标记之一, 广泛应用于种质资源的多样性分析、遗传连锁图谱构建、基因定位及标记辅助育种等研究[ 9]。SSR标记在近缘植物乃至远缘植物基因组间有一定的通用性[ 10]。目前, 许多物种间SSR标记通用性研究已取得较大进展。张扬勇等[ 11]根据拟南芥叶绿体全基因组序列开发89个叶绿体基因组SSR标记, 并分析了标记在甘蓝中的通用性; Wang等[ 12]研究发现禾本科作物小麦、玉米和高粱中的SSR标记在海滨雀稗属作物中的通用性比率分别为67.5%、49.0%和66.8%, 多态性达51.5%。Hu等[ 13]发现黄瓜EST-SSR标记在甜瓜、西瓜、南瓜和葫芦中的具有很高通用性, 通用性比率为92.9%、57.1%、53.6%和60.7%。王丽侠等[ 14]分析187个小豆SSR标记在绿豆中的通用性表明, 约75%的小豆SSR标记可在绿豆中有效扩增。钟敏等[ 15]分析1205个新开发绿豆基因组SSR标记的通用性表明其在豇豆、小豆和饭豆中的通用性比率分别为50.0%、73.3%和81.6%; 多态性比率分别为4.1%、1.7%和1.5%。但与其他物种相比, SSR标记通用性在豆科作物中研究较少, 需进一步探讨不同豆科作物间标记的通用性, 以提高标记的利用效率, 促进遗传研究水平较低的物种的分子遗传学研究。本研究通过分析基于普通菜豆基因组序列开发的SSR标记在豇豆属作物豇豆和小豆中的通用性, 以期增加豇豆、小豆的分子标记, 进而加速豇豆属的基因组学研究。
用于检测普通菜豆SSR标记有效性的普通菜豆地方品种红芸豆(统一编号: F0002322)和京豆(统一编号: F0000777); 用于SSR标记通用性分析的16份小豆材料和16份豇豆材料均由中国农业科学院作物科学研究所提供。16份小豆材料来源于中国11个省市, 16份豇豆材料来源于中国8个省市(表1)。
| 表1 小豆和豇豆品种资源名称及来源 Table 1 Names and origins of adzuki bean and cowpea |
采用天根生化科技(北京)有限公司植物基因组DNA提取试剂盒(离心柱型)提取植物基因组DNA。用1%琼脂糖凝胶电泳检测DNA质量, 紫外分光光度计检测浓度和纯度。
依据普通菜豆基因组序列信息, 利用SSR Locator软件查找SSR位点并设计引物[ 16]。查找SSR位点参数设置: 二核甘酸重复10次, 三核苷酸重复5次。设定引物长度18~22 bp; 引物序列的GC含量40%~60%, 且下游引物GC含量差异小; 引物退火温度在55~60℃之间; 扩增片段长度在100~300 bp之间。引物均由Invitrogen公司合成。
PCR总体积15 μL, 含50 ng基因组DNA, 10×PCR缓冲液1.5 μL (含1.5 mmol L-1MgCl2), 0.25 mmol L-1dNTPs, 上下游引物各0.25 μmol L-1和1 U Taq DNA聚合酶。反应程序为 95℃ 5 min; 95℃ 45 s, 50~60℃ 45 s, 72℃ 45 s, 35个循环; 最后72℃延伸5 min。反应在T100 Thermal Cycler (Bio-Rad Research, USA)扩增仪上进行。SSR扩增产物经6%的聚丙烯酰胺非变性凝胶在60 W恒定功率下电泳 70 min, 用NaOH快速银染法染色后读取条带[ 17]。
利用Popgen32[ 18]软件分析标记多态性信息含量(PIC)、等位基因数( Na)、观察杂合度( Ho)和期望杂合度( He)。
利用SSR Locator软件, 共开发560个菜豆基因组SSR标记。选2份普通菜豆红芸豆和京豆对SSR标记进行初步筛选, 确定有421个SSR标记在菜豆中能够稳定扩增出清晰条带, 有效扩增率为75.2%; 421个有效扩增标记中, 有93个标记在2份材料中表现多态性, 多态性比率为22.1%。
| 表2 普通菜豆基因内SSR标记和基因间序列SSR标记在豇豆和小豆中的通用性和多态性分布 Table 2 Transferability and polymorphism of gene-SSR markers and intergenic region-SSR for cowpea and adzuki bean |
421个普通菜豆SSR标记的基因组序列与菜豆全基因组序列(http://www.phytozome.net/)比对分析显示, 213个标记属于基因内SSR标记, 208个标记属于基因间序列SSR标记(表2)。213个基因内SSR标记分布于普通菜豆11条染色体上, 平均每个染色体上19个标记。Pv03染色体上标记数量最多, 有30个SSR标记, 其次是Pv02和Pv01染色体, 标记数分别为26个和25个。染色体Pv04和Pv05上标记数量最少, 都只有11个(表3)。
| 表3 213个普通菜豆基因序列SSR标记在染色体上的分布 Table 3 Distribution of 213 gene-SSR markers on chromosome in common bean |
分析421个普通菜豆SSR标记在16份豇豆和16份小豆中的通用性显示, 185个菜豆SSR标记在豇豆中能够有效扩增, 标记在豇豆中的通用性比率为43.9%。其中112个标记属于普通菜豆基因内SSR标记, 比率为52.6%; 有73个标记属于普通菜豆基因间序列SSR标记, 比率为35.1% (表2)。161个标记在小豆中能获得有效扩增条带, 标记在小豆中的通用性比率为38.2%。161个通用性标记中, 有95个标记属于普通菜豆基因内SSR标记, 比率为44.6%, 有66个标记属于普通菜豆基因间序列SSR标记, 比率为31.7% (表2)。普通菜豆SSR标记在豇豆属物种豇豆和小豆中通用性存在差异, 在豇豆中的通用性比率高于在小豆中的通用性比率。SSR标记绝大多数在2个物种中能同时得到有效扩增, 2个物种间均能获得有效扩增条带的标记共有138个, 其中基因内的SSR标记达到85个, 基因间序列SSR标记有53个。47个标记只能在豇豆中扩增, 23个标记特异在小豆中扩增。
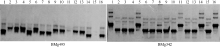
在SSR标记通用性分析的基础之上, 进一步研究了普通菜豆基因组SSR标记在16份豇豆和16份小豆中的多态性(图1和图2)。豇豆中可有效扩增的185个标记中, 有63个标记表现多态, 多态性比率为34.0%, 其中有36个标记属于普通菜豆基因内SSR标记, 比率为32.1%, 27个标记属于普通菜豆基因间序列SSR标记, 比率为37.0% (表2)。PIC值变化在0.110~0.671, 平均PIC值为0.305, 标记的等位基因数量在2~6之间, 平均为2.54; 期望杂合度在0.121~0.706之间, 平均期望杂合度为0.322; 有11个标记位点表现不同程度的杂合性。标记BMg409、BMg497、BMg538、BMg568和BMg569表现较高的杂合度, 观察杂合度均大于0.5, 标记BMg317、BMg445、BMg449、BMg492、BMg567和BMg661观察杂合度小于0.5 (见附表)。
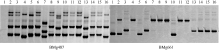
 | 图1 普通菜豆SSR标记在16份豇豆中的扩增图谱Fig. 1 Amplification bands of common bean SSR markers in cowpea |
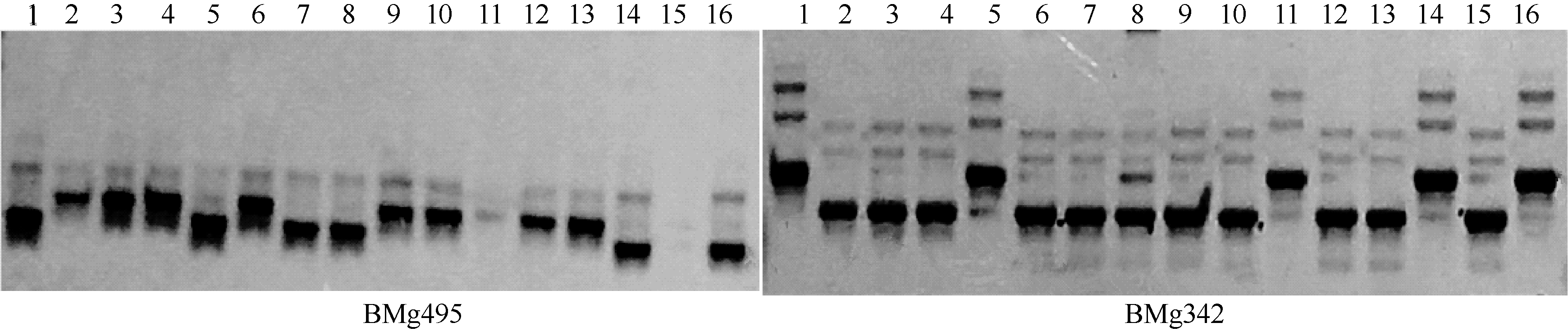 | 图2 普通菜豆SSR标记在16份小豆中的扩增图谱Fig. 2 Amplification bands of common bean SSR markers in adzuki bean |
小豆中有效扩增的161个标记中, 40个标记表现出多态, 多态性比率为24.8%, 其中23个属于普通菜豆基因内SSR标记, 比率为24.2%, 17个属于普通菜豆基因间序列SSR标记, 比率为25.8% (表2)。PIC变化在0.059~0.668, 平均PIC值为0.359, 标记的等位基因数在2~5之间, 平均值为2.8。期望杂合度在0.063~0.734之间, 平均期望杂合度为0.389; 多数多态性标记位点观察杂合度为0, 仅有标记BMg384和BMg445位点观察杂合度大于0, 分别为0.063和0.133 (见附表)。22个普通菜豆SSR标记在豇豆和小豆中存在多态性, 其中13个属于基因内SSR标记, 9个属于基因间序列SSR标记。
分子标记开发的关键是如何高效获得基因组序列或表达的序列标签(EST)序列, 而新一代测序技术可以快速获得海量的序列数据, 极大程度提高了挖掘、验证和评估分子标记的效率[ 19]。小麦[ 20]、水稻[ 21]、甘薯[ 22]、黄瓜[ 23]、西葫芦[ 24]等作物依据基因组序列开发了大量分子标记。豆科作物在这方面的研究也取得一定的进展, Tangphatsornruang等[ 25]利用Roche 454超高通量组测序技术获得470 024条绿豆基因组序列, 并检测到1493个SSR位点用于标记开发。Kaur等[ 26]对小扁豆特异组织进行转录组测序, 获得1.38 ×106个EST, 并开发了2393个小扁豆EST-SSR标记用于后续研究。Garg等[ 27]和Dutta等[ 28]分别利用组装的鹰嘴豆和木豆转录组序列开发了大量功能标记。本研究正是利用Roche 454测序数据, 从中开发SSR标记, 丰富了普通菜豆的遗传标记。
SSR序列在相近的物种间具有一定的保守性, 因而SSR标记在近缘物种间能够通用。标记通用性研究可以为相近物种提供更多的遗传信息, 方便了物种内、种间遗传学与基因组学等方面的研究[ 29]。SSR标记通用性比率高低与所属物种之间亲缘关系密切相关。Gupta等[ 30]随机选取41个豆科模式生物蒺藜状苜蓿SSR标记, 分析显示, 其在豆科植物中的通用性(53%~71%)明显高于非豆科植物(33%~44%)。同科不同属间SSR标记通用性比率明显比同属不同种间通用性低。本研究中普通菜豆与豇豆和小豆是同科不同属作物, 通用性比率为43.9% (豇豆)和38.2% (小豆), 明显低于同属作物绿豆与豇豆和小豆的通用性比率, 后者分别为50.0% (豇豆)和73.3% (小豆)[ 15]。
SSR序列不仅存在于基因序列中, 而且存在于基因间序列中, 由于在物种间基因序列的保守性高于基因间序列的保守性, 因此, 基因内SSR标记在物种间通用性一般高于基因间序列SSR标记。文明富等[ 31]研究木薯419对EST-SSR引物和182对基因组SSR引物在麻疯树和橡胶树通用性发现, 木薯EST-SSR在麻疯树和橡胶树中的通用性比率分别为55.85%和38.90%, 明显高于木薯基因组SSR在麻疯树和橡胶树通用性比率37.36%和26.37%; Varshney等[ 32]分析大麦的EST-SSR和G-SSR标记在近缘物种小麦中的通用性, 发现EST-SSR标记的通用性比率高于G-SSR标记。在本研究中普通菜豆基因内SSR标记在豇豆和小豆中的通用性比率分别为52.6%和35.1%; 高于普通菜豆基因间序列SSR标记在豇豆(44.6%)和小豆(31.7%)中的通用性比率。
多态性分子标记在生物多样性、遗传多样性、物种鉴定等研究中具有广泛的应用价值。标记多态性比率, 多态性标记平均PIC值和平均 He值都可以用来度量群体的遗传多样性。标记多态性比率, 平均PIC值及平均 He值越高, 其群体遗传多样性就越丰富; 反之, 其遗传多样性就越低[ 33, 34]。本研究结果显示普通菜豆基因组SSR标记在16份豇豆和16份小豆中的多态性比率分别为34.0%和24.8%, 多态性标记平均PIC值分别为0.305和0.359, 平均期望杂合度为0.322和0.389, 说明所选用的群体多样性偏低。可能是因为所选取的豇豆和小豆材料数量偏少, 且都是中国栽培种, 起源单一, 材料间的亲缘关系较近。此外, 分子标记多态性高低也与序列所在的位置有一定的关系, 基因内的序列因其保守性较高, 多态性会相对较低, 而基因间序列因其保守性较差, 多态性会相对较高。本文中在豇豆和小豆中基因间标记的多态性都比基因内标记的多态性高。
物种间的遗传关系[ 35]。综合分析SSR标记的通用性和多态性, 可以看出豇豆略高于小豆, 普通菜豆与豇豆的亲缘关系较小豆近。而Sumanasinghe等[ 36]研究表明, 菜豆属的普通菜豆与Ceratotropis亚属小豆的亲缘关系比普通菜豆与 Vigna亚属豇豆的亲缘关系近。本研究中SSR标记基于通用性结果分析的亲缘关系与此正好相反, 主要是因为本研究用于分析的分子标记仅是基因组标记的小部分, 并不能准确地反应物种间基因组信息。只有依据全基因组的序列开发数目更多、类型更加丰富的标记, 才能准确地分析物种的亲缘与进化关系。
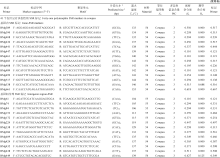
 | 附表 豇豆和小豆中通用性SSR标记信息及在豇豆和小豆中多样性评价Supplemental table Information and diversity asscssments of transferability SSR markers in cowpea and adzuki bean |
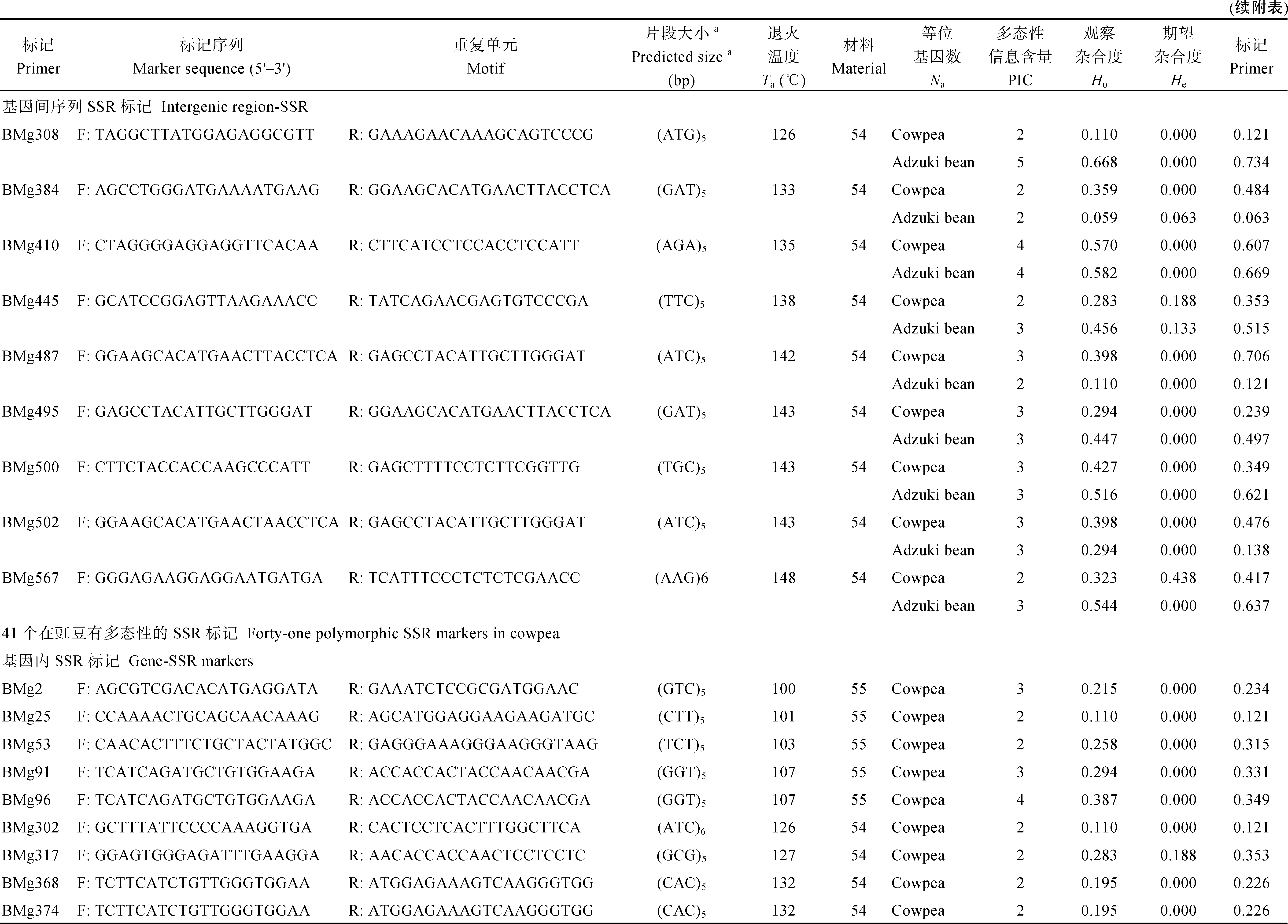 | 附表(续) 豇豆和小豆中通用性SSR标记信息及在豇豆和小豆中多样性评价Supplemental table(Continued) Information and diversity asscssments of transferability SSR markers in cowpea and adzuki bean |
 | 附表(续) 豇豆和小豆中通用性SSR标记信息及在豇豆和小豆中多样性评价Supplemental table(Continued) Information and diversity asscssments of transferability SSR markers in cowpea and adzuki bean |
 | 附表(续) 豇豆和小豆中通用性SSR标记信息及在豇豆和小豆中多样性评价Supplemental table(Continued) Information and diversity asscssments of transferability SSR markers in cowpea and adzuki bean |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|