{kind=link}
{kind=link}
国内外蚕豆核心种质SSR遗传多样性对比及微核心种质构建
[姜俊烨1 , 杨涛1 , 王芳1 , 方俐1 , 仲伟文2 , 关建平1 , 宗绪晓1, *  ]
]
]
|
|


第一作者联系方式: E-mail:jiangjunye123@163.com
运用24对SSR引物, 对国内外1075份初级地理蚕豆核心种质的遗传多样性分析显示, 等位变异数、有效等位变异数及Shannon’s信息指数分别为8.54、2.26和1.02; 对全部参试资源进行聚类分析, 没有发现明显的群体结构, 表明初级地理核心种质的代表性较好, 遗传背景较广泛。之后采用每个类内随机抽样的方法构建含有129份国内资源和63份国外资源的蚕豆微核心种质, 等位基因变异数、有效等位变异数和Shannon’s信息指数的保留比例分别为87.32%、 101.26%和101.82%; 经
The genetic diversity of 1075 genotypes from a primary geographic core collection of faba bean (
蚕豆( Vicia faba L., 2 n=12), 又名佛豆, 胡豆, 罗汉豆等, 是世界上第六大食用豆类作物, 植物学分类上属豆科( Leguminosae), 蝶形花亚科( Papilionoideae), 巢菜属( ViciaL.), 英文名为Faba bean或Broad bean[ 1]。我国已收集保存于种质库中的蚕豆资源近6000份, 数量多, 来源广。中国是全球蚕豆栽培面积及总产量最高的国家, 据FAO[ 2]最新统计资料, 2012年全世界干蚕豆栽培面积243.44万公顷, 总产405.79万吨; 其中中国干蚕豆栽培面积95.30万公顷, 总产140.00万吨, 分别占全世界的39.15%和34.50%。中国每年尚有20~30万公顷的青蚕豆生产面积, 没有列入FAO统计资料。
蚕豆作为小作物, 对其研究进展比较缓慢, 遗传研究基础相对薄弱, 但是随着生物技术的飞速发展, 蚕豆遗传图谱构建取得一定进展。Vande等[ 3]于1991年运用RAPD、RFLP和同工酶等构建了第一张蚕豆遗传连锁图谱, 此图谱有17个标记位于7个遗传连锁群上; 与RAPD、RFLP等分子标记相比, SSR呈共显性, 具有更高的可靠性和重复性, 其两侧具有更保守的序列, 并符合孟德尔遗传定律, 是较适合构建遗传图谱的分子标记[ 4]。Ma等[ 5]于2013年构建了第一张完全基于SSR的遗传连锁图谱, 包含14个遗传连锁群, 127个SSR标记, 这对于蚕豆遗传多样性分析、关联作图、基因定位等均具有极大促进作用。
对于蚕豆遗传多样性的研究, Zong等[ 6]运用10对AFLP分子标记, 对中国国家种质库保存的39份国外资源和204份国内秋播蚕豆资源的遗传多样性进行分析, 共检测出266个多态性扩增片段, 主成分分析和聚类分析显示, 能明显区分开国内外秋播蚕豆; 此外, 王海飞等[ 7]运用11对ISSR标记, 对国家种质库保存的国内外802份蚕豆资源的遗传多样性进行分析, 共扩增出209个多态性片段, 其遗传多样性以中国南方蚕豆较高, 而中国中部地区的蚕豆较低, 通过主成分分析及聚类分析显示, 能明显分开春播蚕豆与秋播蚕豆。
Frankel和Brown[ 8]最先提出核心种质这一概念, 并丰富完善。核心种质是指从整个种质资源中选取一定数量资源作为样本, 以最小的样本数量最大限度地代表整个种质资源的多样性。核心种质的提出为资源的研究提供了一条全新的思路, 而构建核心种质的关键是运用合适的取样方法, 李自超等[ 9]提出, 核心种质的取样比例可以根据原始种质数量而浮动, 原始种质数量较大时核心种质的取样比例可相对较小; 原始种质数量较小时核心种质的取样比例可相对较大。例如, 王丽侠等[ 10, 11]用2794份大豆种质资源为原始材料, 构建占原始种质资源2%的核心种质; 刘勇等[ 12]利用110份柚类构建含原始种质22.7%的核心种质。以上研究结果验证了李自超等[ 9]结论的正确性, 即核心种质数目的选择没有统一化的标准, 要根据原始材料的实际情况来定。微核心种质的代表性可用观测等位基因数, 有效等位变异数以及Shannon’s信息指数的保留比例来确定[ 13]。目前已对很多作物构建了核心种质, 为基因定位和分子标记辅助育种工作提供了便捷途径。Ellis等[ 14]从油菜核心种质中筛选了高抗蚜虫种质, Miklas等[ 15]从普通菜豆核心种质中筛选抗白霉病种质, Santos和Dias[ 16]从甘蓝型油菜核心种质中筛选鉴定出抗白锈病种质等等, 而蚕豆在该方面的研究基本处于空白状态。因此, 本研究从国家种质库保存的6000余份以及美国西部植物引种服务站(WRPIS)提供的蚕豆资源中, 经生态地理筛选获得国内外初级地理核心种质1075份, 旨在运用SSR分子标记对该蚕豆初选地理核心种质进行遗传多样性分析, 进而获得其微核心种质, 从而为目的基因关联分析做铺垫。
1.1.1 参试种质 国家种质中期库中, 有明确地理来源的蚕豆共3620份, 其中国内资源2759份, 国外资源861份。本研究从中选用国内资源716份, 分别来自云南(109份)、浙江(70份)、四川(60份)、江苏(56份)、安徽(56份)、湖北(42份)、甘肃(37份)、湖南(35份)、贵州(35份)、山西(31份)、内蒙古(30份)、陕西(29份)、青海(25份)、重庆(19份)、新疆(18份)、江西(17份)、河北(13份)、宁夏(13份)、广西(11份)、上海(4份)、福建(3份)、广东(1份)的共587个县(区), 每个原产县(县级市)取样1~2份; 国外资源359份, 由美国西部植物引种服务站(WRPIS)提供, 来自全球50个国家。
1.1.2 试剂 SSR引物24对, 参照Ma等[ 5]构建的蚕豆SSR遗传连锁图谱, 分别选自lg1的6对引物SSR6116、SSR10296、SSR10581、SSR9810、SSR2992和SSR5078; 选自lg2的3对引物SSR4076、SSR6092和SSR6221; 选自lg3的4对引物SSR2364、SSR5594、SSR6613和SSR10421; 选自lg4的6对引物SSR102、SSR1929、SSR2577、SSR3581、SSR4798和SSR5740; 选自lg5的3对引物SSR1788、SSR3372和SSR10177; 选自lg8的SSR3637和选自lg10的SSR4056, 按每个连锁群SSR标记数量按比例均匀挑选引物, 能够比较完整地代表蚕豆全基因组。以上引物由北京中科锡林生物科技有限公司合成。 TaqDNA 聚合酶、dNTPs购自北京鼎国昌盛生物技术有限责任公司, DNA marker购自北京泽星生物技术有限责任公司。
1.2.1 DNA提取 从每份参试资源的20个单株上随机选取100~150 mg鲜叶片, 于液氮中冻干研磨成细粉。运用Dellaporta等[ 17]和Doyle等[ 18] 的改良CTAB法提取基因组DNA, 用双蒸水溶解DNA干粉, 置-20℃冰箱备用。
1.2.2 PCR 在PTC-220热循环仪上进PCR, 反应总体积为20 µL, 含DNA (25 ng) 4 µL、10×Tag buffer 2 µL、40 mmol µL-1 dNTPs 0.4 µL、2 µmol µL-1上下游引物各 2 µL、2 U µL-1 Taq DNA聚合酶0.4 µL、ddH2O 11.2 µL。PCR程序为95℃预变性5 min; 95℃变性30 s, 各引物相应退火温度45 s, 72℃延伸45 s, 35个循环; 72℃延伸5 min后于10℃保存。产物经8%非变性聚丙烯酰胺凝胶电泳分离, 银染检测。
1.2.3 数据统计和分析 对每一对参试SSR引物出现的等位变异, 无扩增条带记为0, 其余按分子量由小到大记为1、2、3、……。在NtSYS pc-2.2[ 19]软件包中进行参试资源材料的主成分分析(PCA)和三维作图; 在Structure 2.2.2[ 20]软件包中选择微核心种质数目; 在Popgen 1.32[ 21]软件包中获得群体内等位变异数( Ha)、有效等位基因数( He)、Shannon’s信息指数( I); 用Office Microsoft Excel软件获得 t检验结果。
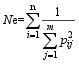

式中, Ne为有效等位基因数, I为Shannon’s信息指数。
24对SSR引物在参试的国内外1075份材料中共扩增出205个等位变异, 平均每对引物扩增出8.5个等位变异, 有效等位变异数为2.26, 其所占比重为26.54%; 24对SSR引物平均Shannon’s信息指数为1.02, 不同的数据指标从不同方面揭示24对蚕豆SSR引物对于揭示1075份国内外蚕豆资源遗传多样性上存在巨大差异。其中, 等位变异数最多的SSR引物是SSR10581, 为25, 其有效等位基因为5.0092; 其次是SSR4798、SSR3372、SSR10421和SSR1929, 等位变异数分别为16、15、11和10, 有效等位
变异分别为2.47、4.36、1.38和2.85; 等位变异数最少的SSR位点是SSR6613, 仅为2, 有效等位变异数为1.83。Shannon’s信息指数( I)最大的位点为SSR10581, 最小的为SSR6092, I值变化范围为0.37~1.97 (表1), 期望杂合度的变化范围为0.17~0.80, 观测杂合度变化范围为0.03~0.98。
| 表1 24对SSR引物在全部参试资源以及微核心种质中扩增的等位变异数、有效变异数和Shannon’s信息指数 Table 1 Alleles, effective alleles, and polymorphic information of SSR amplified products in all tested genotypes and mini-corecollections of faba bean |
数据表明, 1075份蚕豆初级核心资源具有丰富的遗传多样性, 有效等位变异数与Shannon’s信息指数大体呈一致的变化趋势, 有效等位变异数较大的SSR位点其Shannon’s信息指数也较大, 有效等位变异数与等位基因变异频率相关联。与等位变异数相比, Shannon’s信息指数更能反应群体的遗传多样性程度, 是更好的评判指标。
对国内春播、秋播以及国外蚕豆资源分别进行等位变异、有效等位变异和Shannon’s信息指数分析, 得出三者的等位变异数分别为155、162和196, 有效等位变异数分别为52.80、47.14和65.65, Shannon’s信息指数平均数分别为0.94、0.83和1.14 (表2)。
| 表2 24对SSR引物在国内春播、秋播资源以及国外资源扩增的等位变异数、有效等位变异数与Shannon’s信息指数 Table 2 Alleles, effective alleles, and polymorphic information of SSR amplified products among spring, winter sowing accessions from China and accessions from abroad |
数据结果表明, 国外参试蚕豆资源虽然数量比国内参试资源少得多, 但其有效等位变异数却多于国内参试资源; Shannon’s信息指数, 在国外与国内参试蚕豆资源间差异更大。因此, 国外蚕豆资源具有更为丰富的遗传多样性。
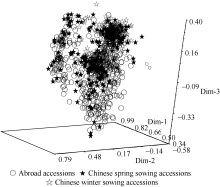
运用NtSYS pc2-2对全部参试蚕豆资源进行三维主成成分(PCA)分析, 得到三维空间聚类图(图1, 前三维贡献率分别为68.88%、6.73%和3.37%)。由图可知, 国外蚕豆资源呈现均匀宽广的空间分布特征, 表明其遗传背景较国内资源更为广泛, 且分布更为均匀; 国内春播、秋播资源混聚在一起, 空间分布范围明显小于国外资源, 说明其遗传背景较国外资源狭窄。另外, 三维图上没有形成依地理来源的资源汇集空间结构, 很可能是因为这些资源已经是经地理筛选得到的初级核心种质, 较好地代表了各种遗传变异而且分布比较均匀, 故而形不成与来源地明显相关的群体结构。
 | 图1 全部蚕豆参试资源PCA分析图Fig. 1 PCA analysis of all accessions |
运用Structure 2.2.2软件包, 将1075份参试资源分为国内和国外两组, 对国内716份资源选取 K=35, 即将国内资源聚类为35个群体, 类内随机选4份资源, 共选取129份资源进入微核心种质(若类内资源数小于4, 则全部进入微核心种质); 对国外359份资源选取 K=35, 类内随机选2份资源, 共选取63份资源(若类内资源数小于2, 则全部进入微核心种质)。所构建的微核心种质(表3)中, 有90份国内秋播资源, 39份国内春播资源以及63份国外资源, 较好地吻合了全部参试资源国内秋播、春播以及国外资源的比例(全部参试资源中, 国内秋播, 春播以及国外资源数分别为518、198和359)。
| 表3 微核心种质编号及来源地 Table 3 Serial number of mini-core germplasm in genebank and their sources |
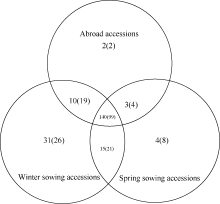
24对SSR引物在构建成的192份微核心种质中共扩增出179个等位变异, 平均每对SSR引物可扩增出7.46个等位变异; 有效等位变异为2.28, 有效等位变异所占比例为30.56%, 24对SSR引物平均Shannon’s信息指数为1.034, 等位变异保留比例 R(%)为87.32%, 有效等位变异数及其所占比重均有所提升(表1), SSR标记期望杂合度变化范围为0.14~0.79, 观测杂合度变化范围为0.05~ 0.98。对初选地理核心种质及其微核心种质的等位变异数、有效等位变异数和Shannon’s信息指数进行t假设检验, 在显著水平为0.05、双尾检验的情况下, 三者的 t值绝对值分别为0.90、0.10和0.15, 均小于 t的双尾临界值2.01, 说明两者差异不显著, 因此基于SSR分子标记数据构建的微核心种质可以代表初级核心种质80%以上的遗传信息。全部参试资源观测等位变异数与微核心种质观测等位变异数对比显示: 全部参试资源中, 观测等位变异在国内春播、秋播以及国外均存在的有140个, 仅春播、秋播和国外资源中独有的观测等位变异分别为2、4和31 (图2); 在构建的微核心种质中则分别为99、2、8和26 (图2)。
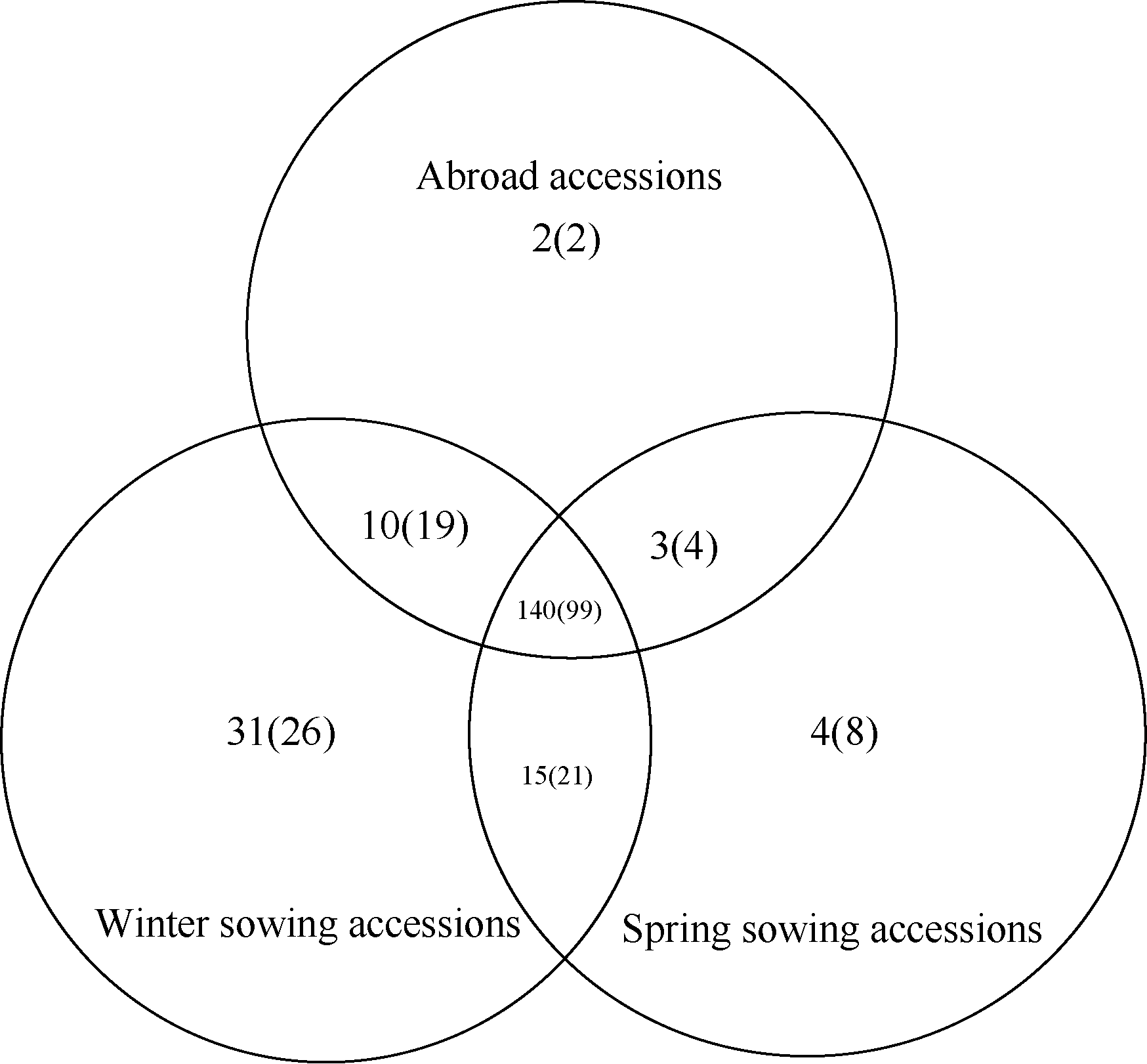 | 图2 全部参试蚕豆资源(微核心种质)等位基因分布图Fig. 2 Distribution of alleles in all resources and mini-core collections |
本研究尝试利用SSR分子标记对全部1075份蚕豆初选地理核心种质进行遗传多样性分析, 并在此基础上构建蚕豆微核心种质, 具有非常重要的理论与实际意义。宗绪晓等[ 6]运用AFLP对国内外243份秋播蚕豆资源进行遗传多样性分析, 共扩增出266个多态性条带; 王海飞等[ 7]运用ISSR对国内外802份蚕豆资源进行遗传多样性分析, 共扩增出209个多态性条带, 上述二人研究材料均由中国农业科学院作物科学研究所提供, 而本研究所选材料不仅包含来自中国农业科学院作物科学研究所716份国内地理核心资源, 还包含美国西部植物引种服务站(WRPIS)提供的359份国外地理核心资源, 较之前研究中所用的蚕豆资源更加全面、丰富; 虽然本研究共扩增出SSR多态性条带204个, 数量稍少于上述研究扩增的, 但是由于SSR分子标记比AFLP、ISSR更为保守, 目标片段所在的遗传连锁群以及在遗传连锁群上的位置明确, 反映的遗传多样性差异更为全面、可信。
所构建的微核心种质, 要以较小的样本数量最大限度地代表整个初选核心种质的遗传多样性, 因此对初选核心种质和微核心种质遗传多样性的评估极为重要。崔艳华等[ 22]通过研究提出, 对于不同的作物, 应该有不同的遗传多样性评估指标和方法。随着生物技术的不断发展, 各种分子标记逐步运用到作物核心种质构建中, 在食用豆核心种质的构建上, 杨菁等[ 23]对153份蚕豆开花天数和株高等18项农艺性状统计分析, 构建了21份青海蚕豆核心种质, 所选核心种质在所有数量特征性状上符合率均占总体资源的88.72%, 21份核心种质的遗传多样性与总体资源差异不显著, 表明其能在形态多样性上代表总体资源; 宗绪晓等[ 24]运用分子标记手段, 对1243份中国地方豌豆资源构建了146份核心种质; 刘玉皎等[ 25]利用AFLP分子标记, 对149份青海蚕豆种质资源进行遗传多样性分析, 构建了40份青海蚕豆核心种质, 包含全部种质80%以上的遗传信息, 可作为杂交育种的优异亲本材料。国内外基于较广范围内的蚕豆资源并未在SSR分子水平上构建微核心种质, 本研究填补这一方面的空白; 运用24对SSR分子标记, 完成国内外1075份蚕豆初级地理核心种质的遗传多样性分析, 并根据Structure 2.2.2软件包聚类结果, 类内随机抽样构建了含192份蚕豆资源的微核心种质, 其中, 中国地方品种129份, 国外品种63份, 占1075份初级核心种质的18.14%, 观测等位变异数, 有效等位基因变异数, Shannon’s信息指数保留比例分别为87.32%、101.26%和101.82%, t检测结果显示微核心种质的遗传多样性与原始地理初选核心种质差异不显著, 证明本研究所构建的微核心种质可以较完整地代替原始地理初选核心种质。
王丽侠等[ 10, 11]提出, 利用SSR数据聚类取样, 在原始种质数目比较大时, 类内随机取样为最佳选择, 本研究证明其结论的正确性。Hintum等[ 26]利用中国大麦地方品种, Balfourier等[ 27]利用饲料作物自然群体, Ortiz等[ 28]利用安第斯山脉的藜麦, Gizlicel等[ 29]利用北美大豆, Malosetti等[ 30]利用乌拉圭地方玉米品种, Chandra等[ 31]利用安第斯山脉四倍体土豆, 对其核心种质的取样策略做了研究。本研究运用王丽侠等[ 10]设计的聚类后类内随机取样法构建蚕豆的微核心种质, 与初级核心种质结果比较证明其可靠性。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|