
{kind=link}
{kind=link}
{kind=link}
等位基因功能差异的统计遗传学分析及应用
胡文明**  , 阚海华
, 阚海华** , 王伟, 徐辰武*
, 阚海华
|
|


* 通讯作者(Corresponding author): 徐辰武, E-mail:qtls@yzu.edu.cn, Tel: 0514-87979358
**同等贡献
等位基因的变异在各种生物中都是普遍存在的, 并对基因的表达起着重要的调控作用。为了探索关联分析中品种数目(A)、平均等位基因多态信息含量(B)和候选基因总贡献率(C)对候选基因分析结果的影响, 本研究采用经验贝叶斯(E-Bayes)方法探讨了上述因素对候选基因检测功效、遗传效应估计值的准确度和精确度以及假阳性出现频率等的影响。结果表明: (1) 随着A、B和C的增加, 候选基因的检测功效和效应估计值的准确度和精确度明显提高, 假阳性出现的频率降低。(2) B对检测功效有显著的影响。在B值保持较高的水平时, 即使品种的数目保持较低的水平以及候选基因的总贡献率较低时, 平均检测功效也可达到80%; 当B值为中等水平时, 需要较大品种数目才能使平均统计功效超过80%; 当B值较小时, 品种数目即使达到100, 3种贡献率水平下的统计功效最高也未达到50%。(3) B对候选基因效应估计值的准确度和精确度有显著的影响。随着B的增加, 候选基因效应估计的准确度和精确度增加。(4) B因素对假阳性频率也有显著影响。在实例分析中检测到4个基因与稻米糊化温度显著关联。因此, 在进行等位基因功能差异的统计遗传学分析时等位基因多态性是主要的影响因素, 同时较多的品种数和较高的贡献率对候选基因的统计功效、效应估计值的准确度和精确度也有重要影响。
**Contributed equally to this work
Allelic variations are ubiquitous in organisms, and play important roles in regulating genes expression. In order to study the influence of number of varieties (A), average polymorphism information content (B) and total contribution of candidate genes (C) on the association analysis of candidate genes, the empirical Bayes (E-Bayes) method was applied to explore the effects of abovementioned three factors on the statistical power of candidate genes, the accuracy and precision of the estimates of genetic effects and the false discovery rate (FDR). Results were as follows: (1) With the increase of factors A, B, and C, the statistical power and the accuracy and precision of the estimates of genetic effects were all enhanced, meanwhile the FDR was decreased. (2) Factor B had a significant influence on the statistical power of candidate genes. When factor B was at a higher level, the ave- raged statistical power could still reach 80% even though both factors A and C remained at lower levels. When factor B was at a medium level, more varieties were needed to ensure that the statistical power could reach 80%. However, when factor B was at a lower level, even though factor A was equal to 100, the statistical power in three different levels of factor C could not reach 50%. (3) Factor B had a significant impact on the accuracy and precision of estimated effects of candidate genes. With the increase of factor B, both the accuracy and precision of effect estimates for candidate genes were improved simultaneously. (4) Factor B also had an important effect on FDR. Through a real data analysis in rice, four detected candidate genes were significantly associated with pasting temperature (PT) by our model. Therefore, the polymorphism information content is a primary factor for detecting the functional difference of alleles. In addition, more varieties and higher contribution rate also have important influence on the statistical power and the accuracy and precision of estimates of effects.
基因是位于染色体上的一段DNA序列, 是生物遗传信息的载体。编码RNA或蛋白质的基因称为结构基因, 不编码RNA或蛋白质的基因称为调控基因。无论是结构基因还是调控基因的序列变异均可对生物体的表型造成影响。等位基因(allele)是位于同源染色体的相同位置上控制某一性状的不同形态的基因, 不同的等位基因各有自己特定的产物和表型。等位基因功能差异最早是在染色体印迹现象中发现的[ 1], 在玉米[ 2]、草莓[ 3, 4]、棉花[ 5]、小麦[ 6, 7]、水稻[ 8]等植物中普遍存在。目前的主要任务是确定更多的等位基因差异与相应的表型多态性之间的联系, 挖掘优异的等位基因和优异的等位基因组合, 构建基因间的互作网络, 使生物学家和遗传学家对生物表型多态性的遗传本质有更深刻的认识。
上述研究虽定性分析了等位基因的差异, 但并未研究基因组中各遗传座位等位基因的联合效应对特定表型的影响。随着全基因组测序技术的日臻成熟以及成本的不断降低, 使得对较多数量基因座位和较多数目品种等位基因序列变异同时检测成为可能, 进而可应用统计学的方法分析等位基因功能差异对表型的影响, 即构建目标性状与候选基因间的遗传模型。其分析方法通常有两类, 第1类是基于连锁分析的QTL定位, 常用的分离群体是双亲杂交衍生, QTL定位方法可以分为标记回归(marker regression, MR)[ 9]、区间作图(interval mapping, IM)[ 10]、复合区间作图(composite interval mapping, CIM)[ 11]、完备复合区间作图(inclusive composite interval mapping, ICIM)[ 12]和多区间作图(multiple interval mapping, MIM)[ 13]等。第2类是基于连锁不平衡理论的关联分析, 分离群体是自然群体, 除了分子标记外, 模型中还包含群体结构或/和亲缘关系协变量。QTL定位软件和关联分析软件, 大多采用逐点扫描, 然后根据一定的测验统计量对每一标记(主效应)或标记组合(互作效应)与表型的关联进行检验。另一种检验表型与标记间关联的方法是同时构建表型值与所有标记间的线性模型, 这类模型中自变量个数远远大于样本数, 属于超饱和模型。常用以下4种方法估计超饱和模型参数: (1)惩罚最大似然方法[ 14](maximum penalized likelihood method, PENAL), 该方法是将所有参数的联合先验分布作为惩罚因子, 与似然函数一起构成惩罚似然函数, 通过最大化惩罚似然函数估计QTL效应。(2)最小绝对缩减和变量选择算子[ 15](least absolute shrinkage and selection operator, LASSO), 是对回归系数加以一定约束条件的最小二乘法。常规最小二乘法(ordinary least square, OLS)对回归系数的估计是通过最小化误差平方和实现的, 但当模型中自变量个数远远大于样本数时, OLS无法估计模型参数。LASSO算法通过施加回归系数绝对值之和小于一个给定的常数约束条件, 从而压缩回归系数, 在此基础上, 再估计OLS参数。(3)随机搜索变量选择法(stochastic search variable selection, SSVS)最初是用来处理线性回归模型的, 后来逐步运用到一些比较复杂的模型, 如广义线性模型[ 16]和对数线性模型[ 17]。2003年, Yi等[ 18]进一步改进了SSVS方法, 使之能够定位多个QTL。(4)逐步回归(stepwise regression method)是多元线性回归分析中较常采用的一种变量选择方法。它的主要思路是将全部自变量按其对依变量作用的显著程度由大到小地逐个引入回归方程, 分为向前选入法(forward)、向后剔除法(backward)和选入与剔除交替进行的逐步回归法(stepwise)。除此之外, 2007年 Xu[ 19]提出一种不依赖于MCMC (markov chain monte carlo)抽样技术的经验贝叶斯方法(E-Bayes), 并成功应用于大麦DH群体有关农艺性状主效应和上位性效应的分析[ 20]。
增加群体大小、减小表型误差、创造近等基因系和染色体片段代换系以及适当增加标记密度都是提高连锁分析检测功效的有效途径[ 21, 22]。本研究将E-Bayes思想用于自然群体等位基因的功能差异分析, 研究品种数目、候选基因的贡献率以及等位基因多态信息含量3个因素对关联分析中候选基因的统计功效、参数估计值的准确度和精确度以及假阳性出现频率的影响, 并进一步探讨其适应条件, 以便为利用种质资源挖掘优异基因提供技术支撑。
设候选基因的数目为 g, 第 i个候选基因具有 ai+1 ( ai≥0)个等位基因, 则该候选基因内的变异具有 ai个自由度, 如以线性模型表示, 相当于 ai个独立变量, g个基因座位共有独立变量 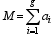
| 表1 第 i个候选基因的 ai+1个等位基因满秩编码 Table 1 Coding for ai independent variables of ai+1 alleles in candidate gene i |
以 yj表示群体中第 j ( j=1, 2, …, n)个品种 nj个个体表型值的平均值, 则 yj具有如下统计遗传模型:
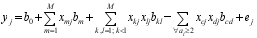
其中, b0表示群体均值, xmj表示第 j个品种在第 m ( m=1, 2, …, M)个变量上的编码值, xkj和 xlj的定义同 xmj; xcj表示第 j个品种在第 i个候选基因座位内的第 c ( c=1, 2, …, ai)个独立变量的编码, xdj定义同 xcj。 bm表示第 m个变量的主效应, bkl表示第 k个独立变量和第 j个独立变量之间的互作效应, bcd表示同一候选基因座位内的 ai个独立变量中第 c个变量和第 d个变量之间的互作效应。由于同一基因座位内独立变量之间的互作效应并不存在, 因此在模型(1)中需要将之删除。例如, 假设有 A、B两个候选基因, 分别具有 A1和 A2以及 B1、 B2和 B3个等位基因, 则 A基因有1个独立变量, 编码为 x1, B基因有2个独立变量, 编码为 x2和 x3, 则(1)式可以表示为:
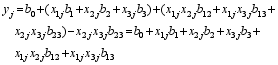
ej为剩余误差, 并假定其遵循平均数为0, 方差为σ2/nj的正态分布。
模型(1)通常是一个超饱和模型, 需采用超饱和模型分析方法统计分析。本文主要采用Xu[ 19]提出的E-Bayes思路估计参数。E-Bayes方法是一种不依赖于MCMC抽样但可以用于超饱和模型参数估计的方法。该方法在进行贝叶斯分析之前, 先运用极大似然估计得到所有的方差分量, 然后将各方差分量作为回归系数先验分布的有关参数, 再利用最优线性无偏估计的方法得到回归系数的后验分布, 最后通过分析后验分布的性质得到参数的估计值。
假设某自然群体包含若干个品种, 为了简化起见并不失一般性, 假定基因组上分布50个候选基因座位, 每个基因座位上只有2个等位基因, 则每个基因座位仅有1个独立变量。共有50×(50+1)/2=1275个可能效应, 其中包含50个主效, 50×(50-1)/2=1225个互作效应。在1275个可能效应中, 随机设置8个遗传效应, 包括主效应4个, 主效应位点间的互作效应1个, 非主效应位点间的互作效应1个, 主效应位点与非主效应位点间的互作效应2个。具体的效应大小和位置设置见表2。
| 表2 单个候选基因的贡献率 Table 2 Contribution of each candidate gene |
共设置3个试验因素, 其中因素A为品种数目, 有4个水平, 分别为A1=30、A2=50、A3=70和A4=100, 每一品种考察的株数为20。因素B为平均多态信息含量, 多态信息含量是供试品种间候选基因座位等位性变异大小的度量。
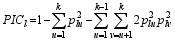
公式(2)中, PICl为第 l座位的PIC值, plu和 plv分别为第 l座位内等位基因 u和等位基因 v的频率, k为该座位等位基因总数, 本研究每一候选基因的 k均为2, 设置等位基因 u的频率为5个水平, 即0.5、0.6、0.7、0.8和0.9, 相应的等位基因 v的频率为0.5、0.4、0.3、0.2和0.1, 用PIC_Calc0.6软件计算PIC值, 由此计算的PIC值共有5个水平, 分别为B1=0.1638、B2=0.2638、B3=0.3318、B4=0.3648和B5=0.3750。因素C为候选基因的总贡献率, 设3个水平, 分别为C1=30%、C2=50%和C3=70%。全试验共4×5×3=60个处理, 每一处理重复模拟100次。贡献率的计算公式见文献[ 19]。
(1)候选基因的统计功效。以100个重复样本中检测到候选基因样本的出现频率来表示。(2)候选基因效应估计值的准确度和精确度。前者以达到显著的若干样本相应候选基因效应估计值的平均值度量, 后者以达到显著的若干样本相应候选基因效应估计值的标准差度量。(3)假阳性候选基因的出现频率。以100个重复样本中出现的所有假阳性候选基因的次数总和表示。
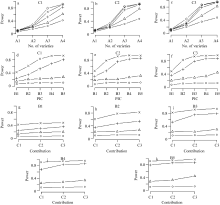
从图1可以看出, (1)随着供试品种数目、PIC和候选基因总贡献率的增大, 候选基因的统计功效呈逐步上升趋势。但不同因素对统计功效的影响大小有别, 影响最大的是A因素, 其次为B因素, 再次为C因素。A因素对统计功效的影响最大, 无论B因素取何种水平, 随着A水平的增加, 统计功效迅速提高, 当A因素取较低的水平时, B因素的各水平间差异不明显, 当A因素取最高水平时, B因素各水平间的差异明显, 尤以B1、B2和B3间的差异最大。(2) B值较高时(例如B5 = 0.375), 品种数目较少(例如70), 候选基因的总贡献率即使较低(例如30%), 平均统计功效也可达到80%; 在此基础上增加品种数目至100统计功效便可增加至90%以上。(3)当B值为中等水平时(例如B3 = 0.3318), 品种数目需要达到100平均统计功效才能超过80%。(4)当B值较小时(例如B1 = 0.1638), 品种数目即使达到100, 3种贡献率水平下的统计功效最高也未达到50%。
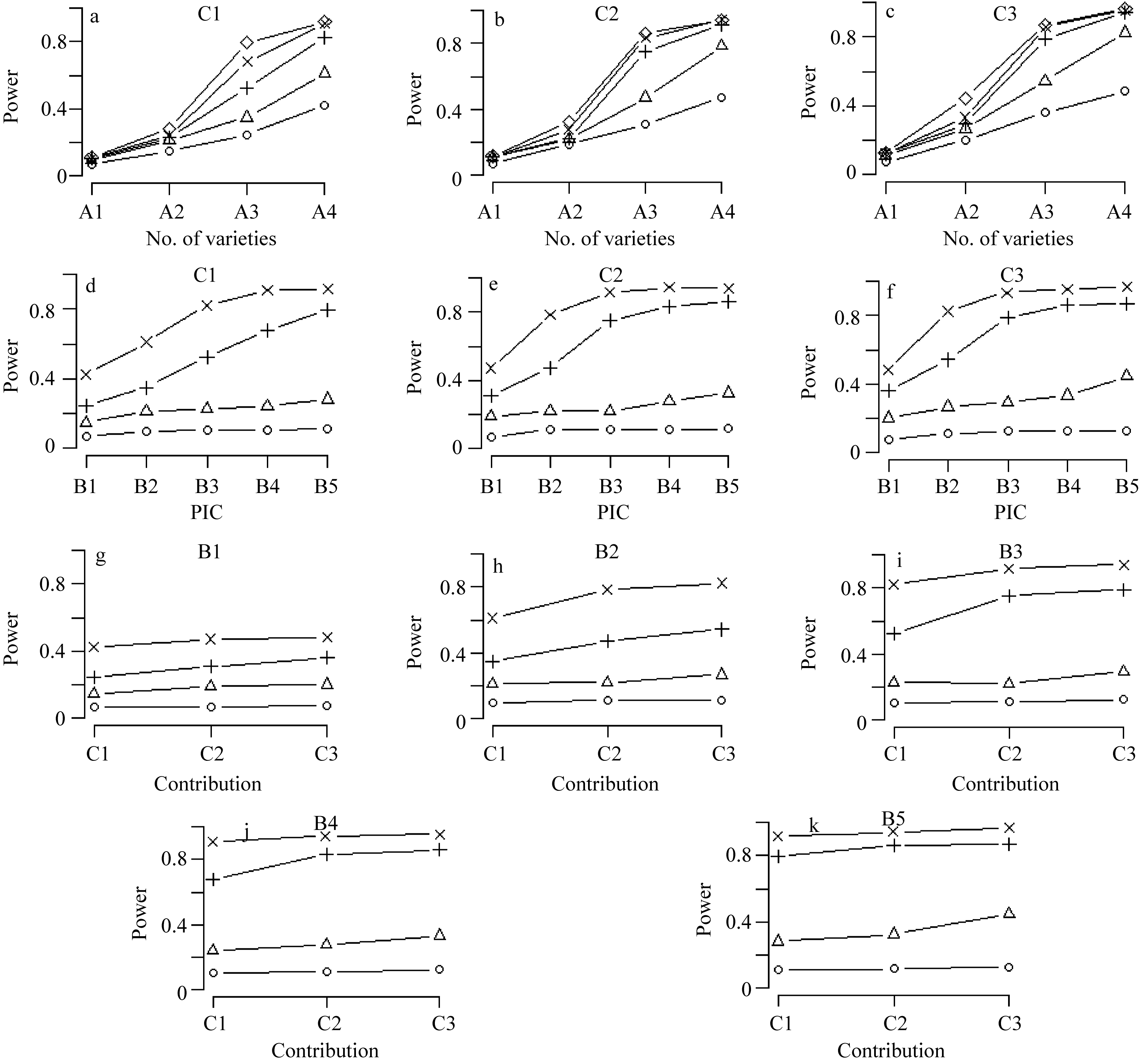 | 图1 品种数目、多态信息含量和候选基因总贡献率对统计功效的影响a~c中○、△、+、×和◇表示B1、B2、B3、B4和B5水平; d~k中○、△、+和×表示A1~A4水平。Fig. 1 Statistic power impacted by number of variants, PIC and total contribution rateIn a-c, ○, △, +, ×, and ◇ represent B1, B2, B3, B4, and B5, respectively; and in d-k, ○, △, +, and ×represent A1-A4, respectively. |
以上结果说明, 在选取品种时要首先保证有足够的品种数目, 其次每一候选基因内的等位变异要尽可能大, 才能有比较好的检测效果, 当品种数较少和等位变异较小, 特别是绝大多数品种都携带同一种等位基因时, 即使该候选基因对表现型有遗传效应, 也不容易被发现。此外, 从本模拟研究可以看出, 只要PIC不太小, 通常70个品种就可保证有较大把握发现中等贡献的候选基因。
从图2可以看出, 候选基因的总贡献率越高, 品种数目越多, PIC值越大, 候选基因的效应估计值愈为准确和精确。但相对而言, PIC值对候选基因的准确度和精确度影响更大, 其次为品种数目, 总贡献率的影响相对较小。例如, 当候选基因的总贡献率为30%时, 候选基因10在同一品种数目水平下随着PIC值的增加其准确度和精确度明显逐步提高。当候选基因的总贡献率为70%时也有同样的变化趋势。但这两种贡献率下候选基因估计值的平均值和标准误却相差不大。
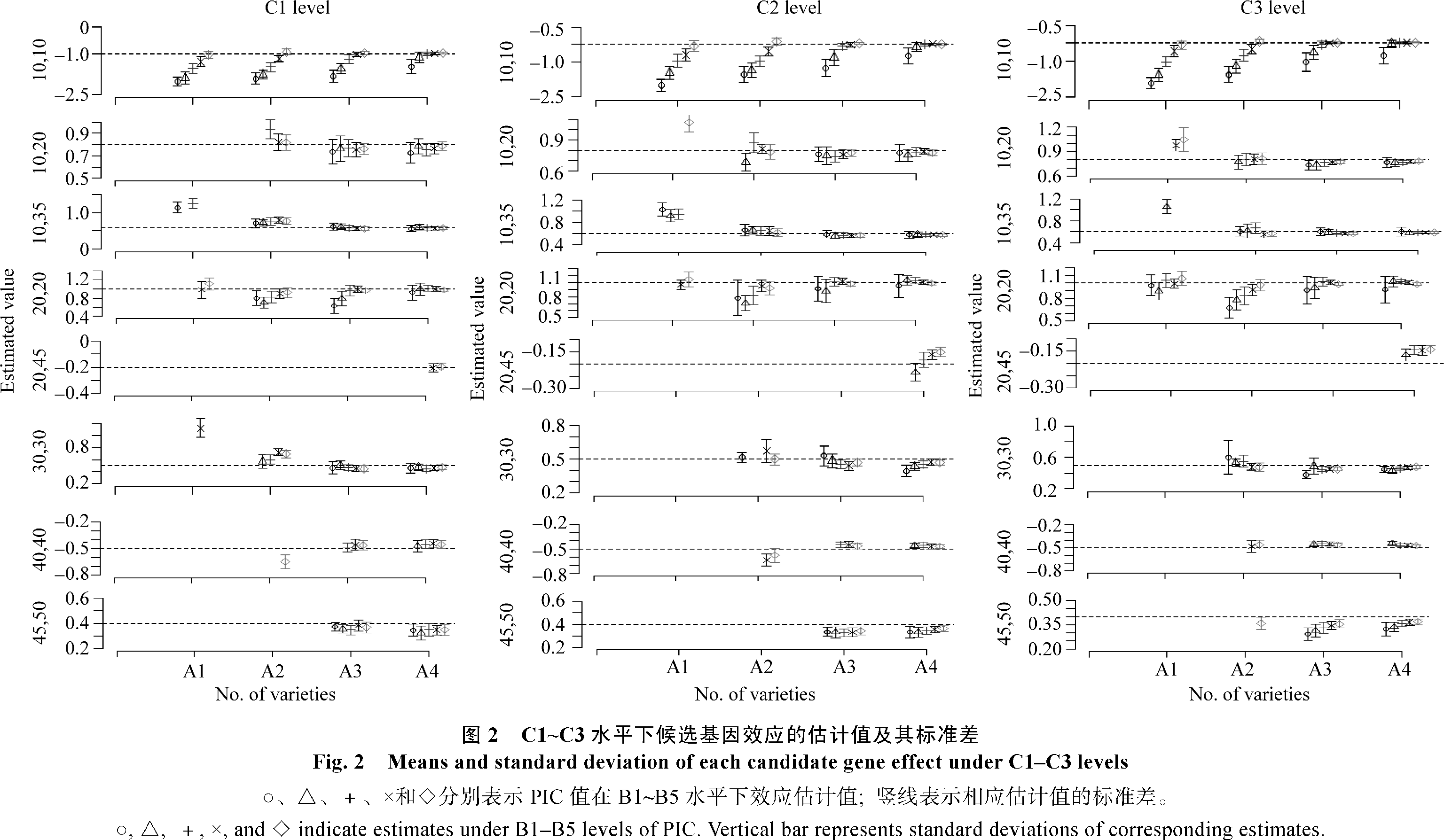 | 图2 C1~C3水平下候选基因效应的估计值及其标准差○、△、+、×和◇表示PIC值在B1~B5水平下效应估计值;竖线表示相应估计值的标准差。Fig.2 Means and standard deviation of each candidate gene effect under C1~C3 levels○, △,+,× and ◇ indicate estimates under B1-B5 levels of PIC. Vertical bar represents standard deviations of corresponding estimates. |
在本研究模拟设置下, 假阳性候选基因只出现在PIC极小的B1和B2两个水平下, 且B1水平下的假阳性候选基因的出现次数明显多于B2水平(表3), 因此, PIC是影响假阳性候选基因出现次数的主要因素。
| 表3 假阳性候选基因的出现次数 Table 3 Frequency of the spurious candidate genes |
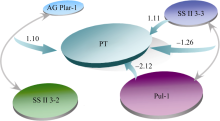
实例数据见文献[8]。供试水稻品种118个。选取在稻米胚乳中表达的与淀粉合成相关的18个基因作为目的基因, 根据日本晴、9311、桂朝2号等13个在品质性状上具有代表性的品种的所有品质相关基因的基因组测序结果, 设计43个分子标记。对以上118个水稻品种进行了基因型检测, 目标性状为RVA黏滞性特性。本文仅以糊化温度(pasting temperature, PT)数据为例分析。利用E-Bayes方法共检测到4个标记与稻米糊化温度显著关联(如图3所示), 分别为标记AGPlar-1、SSII3-2、Pul-1和SSII3-3。其中Pul-1和SSII3-3既有主效应, 又有互作效应; 主效应大小分别为-2.12和1.11, 互作效应大小为-1.26。而AGPlar-1和SSII3-2则仅存在互作效应, 效应大小为1.10。
 | 图3 稻米糊化温度(PT)候选基因及其效应Fig. 3 Analysis result of candidate genes controlling pasting temperature (PT) and their effects |
基因变异在各种生物中是普遍存在的, 其对基因的表达起着重要的调控作用。若变异发生在非编码区, 可以通过增强或减弱该变异位点与结合蛋白的结合, 促进或削弱下游基因的表达, 如果变异发生在编码区, 则该变异的等位基因可能通过编码功能蛋白直接影响表型的大小或编码一种蛋白因子间接地促进或阻碍性状的大小。随着分子生物学研究以及基因组测序技术的发展, 目前我们可以很方便地对有关基因测序从而得到大量序列变异信息, 建立这些变异位点与表型之间的联系已成为目前遗传学研究的热点。本研究设置的可能效应数目与品种数目之比最大为42.5(1275/30), 最小为12.75 (1275/100), 这种可能效应数目远大于品种数目的超饱和模型, 一般可以采用变量选择和压缩估计等方法来处理, Xu[ 19]提出的E-Bayes方法结合了极大似然估计(MLE)和BLUP两种统计方法, 其主要目的是估计回归系数, 而不是方差, 此外E-Bayes方法同时估计所有回归系数的后验平均值, 因此每个回归系数的估计都是独立的, 从而提高了对小样本的估计效率。本文模拟研究了E-Bayes方法, 并取得了较好的分析结果。若采用TASSEL关联分析软件则只能分析基因座位主效应, 无法分析基因座位间的互作效应, 4个主效候选基因的功效分别为94%、93%、67%和57%。而Stepwise是模型选择最直接的一种方法, 运用该方法运算时, 模型收敛的速度比较快, 对于该处理设定的8个效应均能检测到, 统计功效也较高, 但与E-Bayes方法相比, 效应的精确度略低。LASSO和贝叶斯有相似之处, 但其同样不依赖MCMC抽样, 这使得其运算时间大大缩短, 运用LASSO模拟时, 设定的8个效应均能被检测到, 但假阳性较高, 且效应的准确度与精确度均较低。PENAL方法由于其不依赖MCMC抽样, 而是采用极大似然估计的思路, 所以运算速度较快, 但是对于该处理检测功效并不理想, 只检测到4个效应较大的候选基因, 且功效较低, 效应估计值的准确度和精确度较差, 这可能是模型变量远远超过样本容量之故。本研究表明, 候选基因(或基因间的互作)能否被检测到以及被检测到的频率均与该候选基因的贡献率有关, 贡献率越大, 被检测到的概率越高, 例如, 总贡献率为70%时, 贡献率较小的4个效应在品种数目为30时均没有被检测到, 而贡献率最大的候选基因10和20在5种PIC水平下却均能被检测到。本研究构建的模型具有较高的检出率, 当平均等位基因多态性不太小时, 只需要70个个体就能够检测到具有中等贡献率的基因。因此, 进行等位基因功能差异的统计遗传学分析时, 等位基因多态性是主要的影响因素, 同时较多品种的数目和较高的贡献率对候选基因的统计功效、效应估计值的准确度和精确度也有重要影响。
本文提出的模型适用于亲缘关系较远和群体结构不明显的自然群体。利用该模型进行关联分析具有明显的优势, 不仅检出率较高, 而且可以分析基因座位之间的互作。另外, 该模型是对所有的基因座位同时分析, 随机误差降低, 因此, 关联分析的准确度和精确度都较高。而常用的QTL定位方法和关联分析方法一般是对单个基因座位逐个检测, 导致误差效应与大量的微效多基因效应混合, 每次检测时误差不统一, 准确度和精确度相对较低。因此, 本文基于全基因组所有座位进行变量筛选的模型选择方法具有较高的利用价值。下一步的工作设想是对模型作进一步改进, 考虑消除群体结构和亲缘关系对分析结果的影响, 使之适用于任何的自然群体, 进一步提高模型的适用范围。
进行等位基因功能差异的统计遗传学分析时, 等位基因多态性信息含量是主要的影响因素, 同时较多品种的数目和较高的贡献率对候选基因的统计功效、效应估计值的准确度和精确度也有重要影响。
| [1s] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|