



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
不同连作年限野生地黄根际土壤微生物群落多样性分析
[吴林坤1, 2  , 黄伟民
, 黄伟民1, 2, * , 王娟英1, 2, * , 吴红淼1, 2 , 陈军1, 2 , 秦贤金1, 2 , 张重义2 , 林文雄1, 2, * ]
, 黄伟民]
|
|


第一作者联系方式: E-mail: wulinkun619@163.com **同等贡献(Contributed equally to this work)
以野生地黄为试验材料, 设置野生地黄头茬土壤、重茬土壤和原茬土壤处理, 未种植任何作物为对照, 于块根膨大中期采集土样, 通过磷脂脂肪酸法(PLFA)和末端限制性片段长度多态性(T-RFLP)技术, 分析不同连作年限野生地黄的根际微生物生物量和群落结构变化。PLFA分析结果表明, 不同处理情况下地黄根际土壤微生物群落结构存在明显差异, 与头茬地黄根际土壤相比, 重茬地黄土壤微生物总量显著下降, 并且细菌/真菌比例下降。T-RFLP分析结果表明, 不同连作年限地黄根际土壤细菌群落结构存在一定差异, 野生状态地黄土壤和头茬土壤菌群较为相似, 变形菌门和厚壁菌门占据优势地位。野生状态地黄和头茬地黄根际富含 Bacillus、 Pseudomonas等有益生防菌, 而重茬地黄根际土壤滋生大量病原菌如 Clostridium sp.、 Flexibacter polymorphus和 Clostridium ghoni, 有益菌群和纤维素降解菌群减少, qRT-PCR定量分析也显示, 野生状态地黄和头茬地黄土壤中假单胞菌数量都显著高于重茬地黄土壤。总之, 野生地黄存在连作障碍问题, 导致野生地黄根际有益菌数量减少而病原菌大量滋生, 从而降低了野生地黄抵御病害的能力, 使重茬野生地黄生长发育差, 产量大幅降低。
, HUANG Wei-Min
The soils sampled from the four different plots, including the newly planted, the two-year monocultured, the wild R. glutinosa and the control without growing R. glutinosa, were used to study the changes in microbial biomass and community composition using phospholipid fatty acid (PLFA) and terminal restriction fragment length polymorphism (T-RFLP) analyses. PLFA analysis indicated that the soil microbial community composition was significantly different among the R. glutinosa with different years of monoculture. Compared with the newly planted soil, the total PLFA content and the ratio of bacteria/fungi in two-year monocultured soil greatly declined. Further analysis by T-RFLP also displayed the distinct differences in rhizospheric bacterial community structure of R. glutinosa. The microbial compositions from the wild and the newly planted R. glutinosa soils tended to be more similar. It was found that the bacteria including Proteobacteria and Firmicutes were predominant in the wild and newly planted R. glutinosa soils. Some beneficial biocontrol bacteria (such as Bacillus, Pseudomonas, etc.) gathered in the rhizosphere of the wild and newly planted R. glutinosa. However, a large number of pathogenic bacteria bred in the rhizosphere of the two-year monocultured R. glutinosa, such as Clostridium sp., Flexibacter polymorphus, and Clostridium ghoni, and the number of beneficial bacteria and cellulose degradation bacteria decreased. Furthermore, qRT-PCR analysis verified that the total number of Pseudomonas was much higher in the wild and newly planted R. glutinosa soils than in the two-year monocultured soil. In conclusion, the pathogenic microbes breed seriously in the rhizospheric soil of wild R. glutinosa under the monoculture regime, and yet the number of beneficial bacteria decline, resulting in weakened ability of wild R. glutinosa to resist the diseases so that the two-year monocultured wild R. glutinosagrows abnormally and its yield is decreased drastically.
地黄(Rehmannia glutinosa Libosch)是一种重要的常用传统补益中药, 始载于《神农本草经》, 其块根经各种炮制过程后即可入药, 具有多种功效, 如滋阴补血、清热生津、益精填髓等, 是我国常用的大宗药材之一。但是, 地黄在栽培过程中存在明显的连作障碍问题, 造成产量品质严重下降, 甚至绝收, 每茬种植后需间隔8~10年方可再种[1], 这限制了中药资源的可持续生产以及地黄产区区域经济的快速发展。
微生物与土壤生态功能是相辅相成、息息相关的, 并且跟植物之间有着紧密的联系, 因此, 微生物群落的组成及其多样性是衡量土壤性质和功能的一个重要指标。连作障碍形成及加重的原因极其复杂, 根际作为植物-土壤生态系统物质交换的一个界面, 是根系-土壤-微生物三者紧密结合、相互交流的场所[2]。植物可以通过根系分泌某些化合物来吸引某些特定微生物类群, 也能选择性抑制某些微生物类群[3]。反之, 微生物类群的数量和多样性的改变, 也会影响土壤的微生态功能[4]。Mendes等[5]分析抑病型土壤(disease-suppressive soil)与利病型土壤(disease- conducive soil)的微生物群落结构发现, 种植于抑病型土壤中的甜菜其根际优势群落主要为一些有益拮抗菌(如β -变形菌门、γ -变形菌门、放线菌门等), 尤其发现抑病型土壤中的假单胞菌数量极显著高于利病型土壤。王明道等[6]运用变性梯度凝胶电泳和传统的平板培养法检测地黄连作过程中土壤微生物类群的变化, 发现连作导致细菌数量减少, 木霉和黄曲霉数量增加, 土壤生态系统失调。Van Bruggen等[7]研究发现根际微生物的分布状态与沿根尖的分泌物含量有密切关系, 微生物生物量的积累在一定程度上依赖于根系分泌物的释放。因此, 分析土壤微生物群落结构及其多样性和动态性等信息, 对深入系统地揭示植物连作障碍的机制具有重要意义。
众所周知, 传统的微生物培养方法受诸多因素制约, 这不仅是由于自然界中可培养的微生物只有约0.01%~10.00%, 而且通过分离得到的微生物不能准确地反映自然环境中此物种的原位信息。因此, 通过分子生物学技术研究土壤微生物的群落和功能多样性成为一种必然的趋势。近年来, 随着生物学的发展, 相继出现了许多用于土壤微生物群落结构分析的研究技术, 如限制性片段长度多态性(RFLP)、磷脂脂肪酸(PLFA)、变性梯度凝胶电泳(DGGE)和末端限制性片段长度多态性(T-RFLP)技术等[8]。目前, T-RFLP技术在分析比较不同土壤样品中微生物群落的多样性以及探讨不同土壤微生物间的系统发育和亲缘关系方面发挥着重要作用[9, 10, 11]。
本研究结合运用磷脂脂肪酸法(PLFA)和末端限制性片段长度多态性(T-RFLP)技术分析和比较不同连作年限的野生地黄及野生状态地黄根际微生物群落结构和多样性变化, 以期为深入揭示药用植物连作障碍形成的分子生态机制和探索合理有效的消减策略提供理论依据。
将野生地黄人工移栽至河南农业大学试验农场大田连续种植2年, 共设置以下4个试验, 即头茬土壤(ZC, 移栽至大田种植1年)、重茬土壤(CC, 移栽至大田连续种植2年)、野生状态地黄土壤(WR)以及对照土壤(CK, 空白土壤即整个试验过程中未种植任何作物)。其中, 野生状态地黄土壤采用5点取样法(WR), 取样地点土壤质地、气候等条件与田间试验基本相同, 野生状态地黄周围其他植被多样性丰富(野生地黄占总植被比例平均为26.2%)。本试验所用地块(正茬、重茬、对照)均为新开垦的山地土壤, 在同一地块上, 肥力水平一致。整地种植前, 基施三元复合肥750 kg hm-2, 磷酸二胺550 kg hm-2, 过磷酸钙750 kg hm-2, 硫酸钾复合肥375 kg hm-2; 地黄块茎膨大时追施三元复合肥225 kg hm-2。整个试验过程中, 头茬、重茬以及对照地块的田间管理措施(如施肥和水分管理等)均保持一致, 野生状态地黄未进行施肥除草等农艺措施, 保持自然野生状态。
于块根膨大中期采用抖根法采集土样, 即用铲子挖出完整地黄块根, 用力抖动去除块根表面疏松土壤, 再用刷子刷取紧贴地黄块根表面的土壤(50 g 株-1)作为根际土壤。过筛后将土壤样品分成2份, 一份自然风干后用于土壤理化性质测定; 一份迅速置-80℃冰箱或立即用于土壤微生物磷脂脂肪酸和土壤总DNA提取。
参照文献[12], 采用重铬酸钾氧化法测定有机质; 采用碱解扩散法测定碱解氮; 采用0.5 mol L-1 NaHCO3浸提-钼锑抗比色法测定速效磷; 采用0.5 mol L-1 NH4OAc浸提-火焰光度法测定速效钾; 采用pH酸度计(Sartorius PB-10)测定pH值。
采用磷脂脂肪酸(PLFA)法测定不同连作年限地黄根际土壤微生物量。以KOH-甲醇溶液甲酯化法提取磷脂脂肪酸[13, 14], 以十九烷酸甲酯(19:0)为内标。微生物特征性脂肪酸分析中, 细菌标记性脂肪酸为15:0、16:1w7c、a17:0、cy19:0等[15, 16]; 真菌标记性脂肪酸为18:1w9c、18:2w6t[13, 17]; 放线菌标记性脂肪酸为10Me17:0、10Me18:0[13, 18]; 原生动物标记性脂肪酸为20:4w6c(6, 9, 12, 15)[19]。其中, i、a、cy和Me分别表示同型(甲基在C末端第2位碳原子上)、异型(甲基在C末端第3位碳原子上)、环丙基和甲基分支脂肪酸; w后的数字表示双键的位置(甲基端起); c、t分别表示顺式(双键两侧-H在同侧)及反式(双键两侧-H在异侧)脂肪酸。
参照文献[20]并稍作修改提取土壤微生物总DNA。用1%琼脂糖凝胶检测DNA, 用胶回收试剂盒(TianGen Biotech Co., Ltd)纯化DNA, 纯化后置-20℃冰箱保存, 每个土样均3个重复。
采用细菌通用引物扩增细菌16S rRNA特异片段, 引物为8-27F (5° -AGAGTTTGATCCTGGCTCA G-3° )和1492R (5° -TACGGTACCTTGTTACGACTT- 3° ), 其中8-27F引物的5° 端用FAM标记荧光[21]。PCR反应体系(25 μ L)为12.5 μ L 2× Taq混合物(Sangon Biotech Co., Ltd.), 1.0 μ L 8-27F引物, 1.0 μ L 1492R引物, 1.0 μ L DNA模板, 加9.5 μ L ddH2O。PCR反应扩增程序为 94℃预变性5 min; 94℃变性1 min, 55℃退火50 s, 72℃延伸100 s, 35个循环, 最后72℃延伸10 min, 4℃保存。扩增产物经胶回收试剂盒(TianGen Biotech Co., Ltd.)回收后用1%琼脂糖凝胶电泳检测。纯化的PCR产物用Hae III、Msp I、Afa I和Alu I限制性内切酶进行酶切[22]。
酶切产物脱盐后与上样缓冲液及标准Marker (GeneScan-500, Applied Biosystems)混合均匀, 96℃变性4 min, 迅速置冰上, 然后在ABI 3730xl DNA sequencer测序分析仪(Applied Biosystems)上进行毛细管电泳检测。
测序结果采用GeneMarker V1.2进行图谱分析, 进一步, 将4个限制性内切酶(Msp I、Hae III、Afa I和Alu I)酶切产生的T-RF片段与RDP数据库进行比对, 找出各样品中每个T-RF片段所对应的微生物种属。
假单胞菌(Pseudomonas)定量分析所用引物为Ps-for (5'-GGTCTGAGAGGATGATCAGT-3')和Ps-rev (5'-TTAGCTCCACCTCGCGGC-3')[23]。PCR反应体系含12.5 μ L 2× Taq混合物(Sangon Biotech Co., Ltd.)、引物各1.0 μ L、1.0 μ L DNA模板, 加9.5 μ L ddH2O, 总体积为25 μ L。PCR扩增程序为94℃预变性5 min, 95℃变性1 min, 64℃退火1 min, 72℃扩增1 min, 35个循环, 最后72℃再继续延伸10 min, 4℃保存。
采用Microsoft Excel 2003分析数据, 方差分析和聚类分析采用DPS7.0软件, 主成分分析(Principal Component Analysis)采用SPSS11.0软件。
头茬野生地黄和重茬野生地黄在长势上存在显著差异, 头茬野生地黄长势旺盛, 地上部植株健壮、叶片宽大, 地下部块根膨大正常(图1-A、图1-E左侧), 与林子中野生状态地黄长势相近(图1-C), 且无病害症状; 而重茬野生地黄植株矮小, 叶片小, 地下部块根无法伸长生长和膨大, 并出现明显的枯萎死亡现象(图1-B、图1-E右侧); 野生状态地黄地上部植株长势良好, 块根也能正常膨大(图1-C、图1-D)。
 | 图1 地黄田间生长情况A: 正茬野生地黄; B: 重茬野生地黄; C: 野生状态地黄; D: 野生状态地黄地下部; E: 正茬(左)、重茬(右)野生地黄地下部。Fig. 1 Growth status of R. glutinosa in the fieldA: newly plantedR. glutinosa; B: two-year monoculturedR. glutinosa; C: wild R. glutinosa; D: tuber roots of the wild R. glutinosa; E: tuber roots of the newly planted (left) and two-year monocultured (right)R. glutinosa. |
从表1可以看出, 不同连作年限地黄根际土壤中速效养分存在显著性差异, 野生状态地黄根际土壤的速效养分均最低, 这可能与野生状态下未施肥以及周围其他植被共同吸收利用有关, 而重茬土壤样品中碱解氮、速效磷、速效钾的含量分别为54.36, 59.75和21.28 mg kg-1, 都显著高于头茬和野生状态地黄根际土壤, 可见地黄连作并未导致土壤速效养分下降, 反而有积累效应。同时, 发现重茬野生地黄土壤的pH值显著低于头茬和野生状态地黄根际土壤。
| 表1 不同连作年限地黄根际土壤化学性质比较 Table 1 Chemical properties of R. glutinosa rhizospheric soils with different year monocultures |
经GC-MS测定, 共鉴定出19种微生物标记性脂肪酸(表2)。野生状态地黄、头茬地黄、重茬地黄根际土壤以及空白对照土壤的微生物总量分别为62.255、54.848、48.398和51.565 nmol g-1(表3), 可见重茬地黄根际土壤微生物总量与头茬相比显著下降。
4种不同连作年限地黄其根际土壤微生物群落特征差异显著(表2)。其中, 15:0、16:1w7c、a17:0、cy19:0等表示细菌; 18:1w9c、18:2w6t表示真菌; 10Me17:0、10Me18:0表示放线菌; 20:4w6c (6, 9, 12, 15)表示原生动物。由表3可以看出, 野生状态地黄根际土壤中真菌和细菌的含量都较高, 这可能与野外生态系统地黄周围植被较为丰富有关, 微生物群落达到一个相对的平衡。比较发现, 重茬土壤微生物群落中细菌含量下降, 而真菌含量却上升, 即细菌/真菌比例下降。
| 表2 不同连作年限地黄根际土壤中各种磷脂脂肪酸含量 Table 2 Concentrations of phospholipid fatty acids in R. glutinosa rhizospheric soils with different year monocultures (nmol g-1soil) |
| 表3 不同连作年限地黄根际土壤微生物量 Table 3 Microbial biomass in R. glutinosa rhizospheric soils with different year monocultures (nmol g-1) |
2.4.1 不同连作年限地黄根际土壤微生物DNA提取及PCR扩增 由图2-A可以看出, 以CTAB法提取的土壤总DNA条带较为清晰, 纯度较高。以Universal DNA Purification Kit胶回收试剂盒(TianGen Biotech Co., Ltd)回收后的DNA作为模板进行细菌16S rRNA扩增, 回收后检测发现, PCR扩增产物为1500 bp左右, 且片段特异性良好(图2-B)。
 | 图2 地黄根际土壤微生物总DNA(A)及16S rRNA扩增产物(B)CK: 对照土; ZC: 头茬土壤; WR: 野生土壤; CC: 重茬土壤。Fig. 2 Electrophoresis results of soil DNA (A) and the PCR products of 16S rRNA (B)CK: control soil; ZC: newly planted soil; WR: wildR. glutinosa soil; CC: two-year monocultured soil. |
2.4.2 不同连作年限地黄根际土壤细菌T-RFLP图谱分析 将扩增的16S rRNA产物分别用4种限制性内切酶(Msp I、Hae III、Afa I和Alu I)进行酶切, 在野生状态地黄土壤中分别检测到T-RF片段155、141、80和140个; 在头茬地黄土壤中分别检测到T-RF片段137、128、152和109个; 在重茬地黄土壤中分别检测到T-RF片段114、122、144和129个; 在对照土壤中分别检测到T-RF片段127、123、86和165个(图3)。可见, 不同连作年限的地黄根际土壤中细菌群落结构存在一定差异。
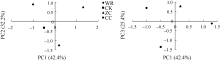
2.4.3 不同连作年限地黄根际土壤细菌群落结构分析 根据Msp I酶切片段的相对含量, 对不同连作年限地黄根际土壤细菌群落结构进行主成分分析, 主成分1、主成分2、主成分3分别解释变量方差的42.4%、32.2%、25.4%, 三者共解释了变量方差的100.0%。由图4可见, 主成分1、主成分2、主成分3能够有效区分不同连作年限地黄根际土壤的细菌群落, 其中野生状态地黄和头茬地黄均处于主成分1的正半轴, 说明野生状态地黄和头茬地黄根际土壤的细菌群落结构较为接近, 而重茬地黄土壤与其他3个样品相比, 区别较为明显, 处于主成分1负半轴的远端, 说明地黄连作明显改变了根际土壤细菌的群落结构。聚类分析也表明, 重茬地黄根际土壤细菌群落结构与对照土壤最为接近, 而明显区别于头茬地黄和野生状态地黄(图5)。
 | 图3 不同连作年限地黄根际土壤细菌群落的酶切图谱CK: 对照土; ZC: 头茬土壤; WR: 野生土壤; CC: 重茬土壤。Fig. 3 T-RFLP profiles of bacterial community in rhizospheric soils of R. glutinosa with different year monoculturesCK: control soil; ZC: newly planted soil; WR: wildR. glutinosa soil; CC: two-year monocultured soil. |
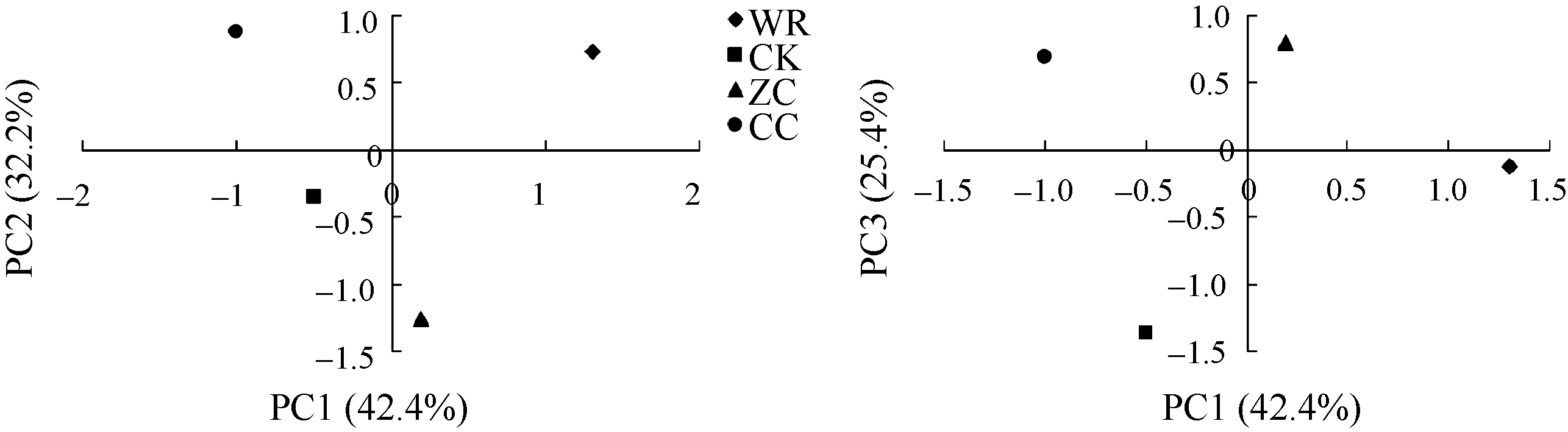 | 图4 不同连作年限地黄根际土壤细菌群落结构的主成分分析CK: 对照土; ZC: 头茬土壤; WR: 野生土壤; CC: 重茬土壤。Fig. 4 Principal component analysis of rhizospheric bacterial community of R. glutinosa with different year monoculturesCK: control soil; ZC: newly planted soil; WR: wildR. glutinosa soil; CC: two-year monoculture soil. |
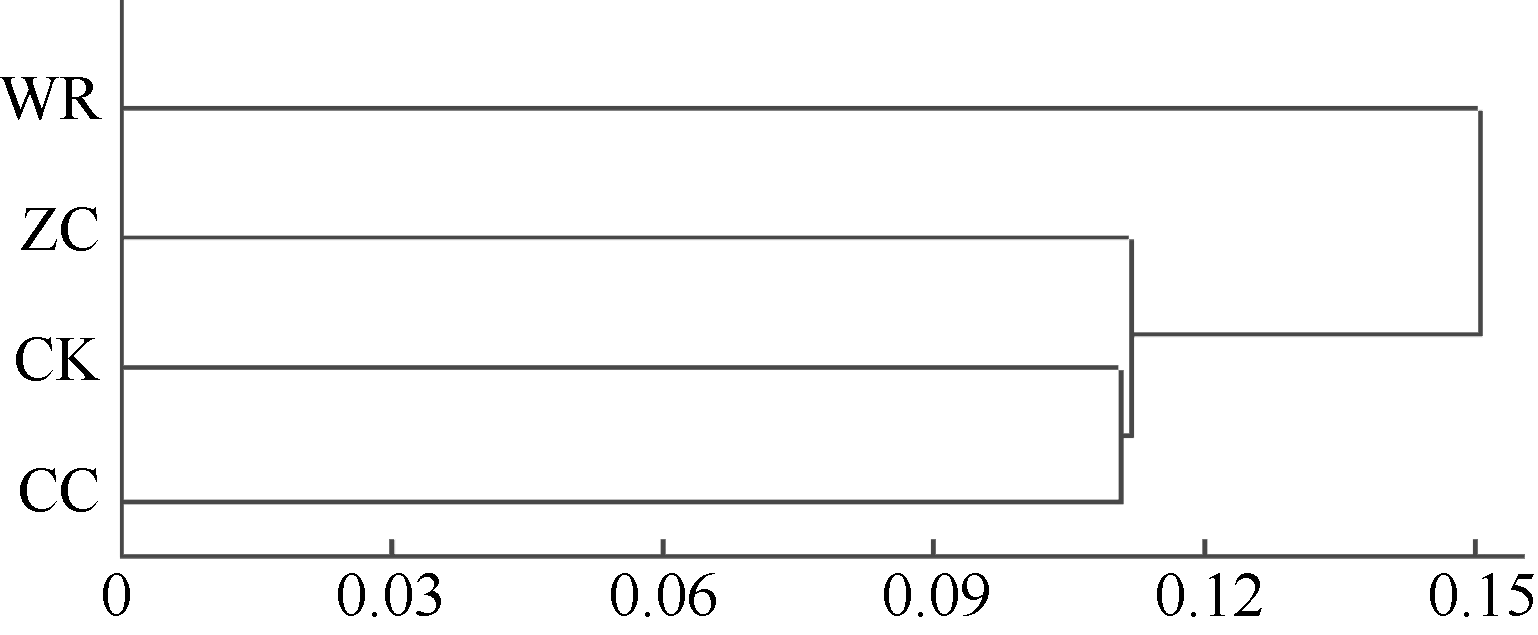 | 图5 不同连作年限地黄根际土壤细菌群落结构的聚类分析CK: 对照土; ZC: 头茬土壤; WR: 野生土壤; CC: 重茬土壤。Fig. 5 Clustering analysis of rhizospheric bacterial community of R. glutinosa with different year monoculturesCK: control soil; ZC: newly planted soil; WR: wildR. glutinosa soil; CC: two-year monocultured soil. |
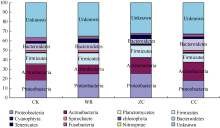
 | 图6 不同连作年限地黄根际土壤细菌群落结构分析CK: 对照土; ZC: 头茬土壤; WR: 野生土壤; CC: 重茬土壤。Fig. 6 Bacterial community structure in rhizospheric soils of R. glutinosa with different year monoculturesCK: control soil; ZC: newly planted soil; WR: wildR. glutinosa soil; CC: two-year monocultured soil. |
通过对不同连作年限地黄根际微生物进行门纲分类分析后发现, 变形菌门的细菌种类在野生状态地黄和头茬地黄根际土壤中所占比例分别为25.08%和25.42%, 均高于重茬地黄的18.16%, 厚壁菌门在野生状态地黄(12.54%)和头茬地黄(12.71%)根际土壤中所占的比例也大于重茬地黄的9.20%。然而, 放线菌菌门在野生状态地黄、头茬地黄、重茬地黄根际土壤中的比例分别为9.48%、16.72%和19.34%, 即呈递增的趋势。
2.4.4 不同连作年限地黄根际土壤细菌群落的功能分析 将4种限制性内切酶酶切产生的T-RF片段与RDP数据库比对, 并对各处理的一些优势菌群进行功能归类分析, 共分为5大类, 即碳循环、硫循环、纤维降解菌、有益菌以及病原菌(表4)。其中, 碳循环功能菌有拟杆菌属、醋杆菌属、嗜甲基菌属3种; 硫循环功能菌有硫化杆菌属2种; 纤维降解菌有噬纤维菌属2种。整体来看, 多数碳循环、硫循环或纤维素降解相关的细菌菌群, 在野生状态地黄和头茬地黄根际土壤中的相对含量都明显高于重茬地黄土壤。可见, 地黄连作导致一些参与土壤营养循环的微生物数量下降。有益菌有芽孢杆菌属、类芽孢杆菌属、假单胞菌属3种。其中芽孢杆菌属在野生状态地黄、头茬地黄、重茬地黄、对照土壤中的相对含量分别为4.56%、4.89%、1.17%和1.62%; 假单胞菌属在野生状态地黄、头茬地黄、重茬地黄、对照土壤中的相对含量分别为1.03%、1.13%、0和0。病原菌主要有梭菌属2种和屈挠杆菌属1种。其中Clostridium sp.在野生状态地黄、头茬地黄、重茬地黄、对照土壤中的相对含量分别为0.79%、0.76%、2.00%和0.42%; Flexibacter polymorphus在野生状态地黄、头茬地黄、重茬地黄、对照土壤中的相对含量分别为1.57%、1.71%、2.92%和1.64%; Clostridium ghoni在野生状态地黄、头茬地黄、重茬地黄、对照土壤中的相对含量分别为0.67%、0.94%、1.94%和1.17%。几类有益菌含量变化都呈现重茬地黄土壤低于野生状态地黄和头茬地黄土壤的趋势, 而几类潜在病原菌的含量变化都呈现出重茬地黄土壤高于野生状态地黄和头茬地黄土壤的趋势。
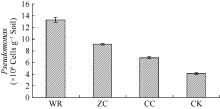
2.4.5 不同连作年限地黄根际土壤中假单胞菌qRT-PCR定量分析 基于T-RFLP的分析结果, 确定了一类潜在的有益菌群即假单胞菌。进一步采用qRT-PCR技术对不同连作年限地黄根际土壤中假单胞菌含量进行绝对定量分析, 发现野生状态地黄和头茬地黄根际土壤中假单胞菌含量显著高于重茬地黄土壤, 这与T-RFLP分析结果相一致, 验证了T-RFLP结果的准确性。
| 表4 不同连作年限地黄根际土壤特异菌群相对含量分析 Table 4 Changes in the relative abundance of specific bacteria in rhizospheric soils of R. glutinosa with different monoculture |
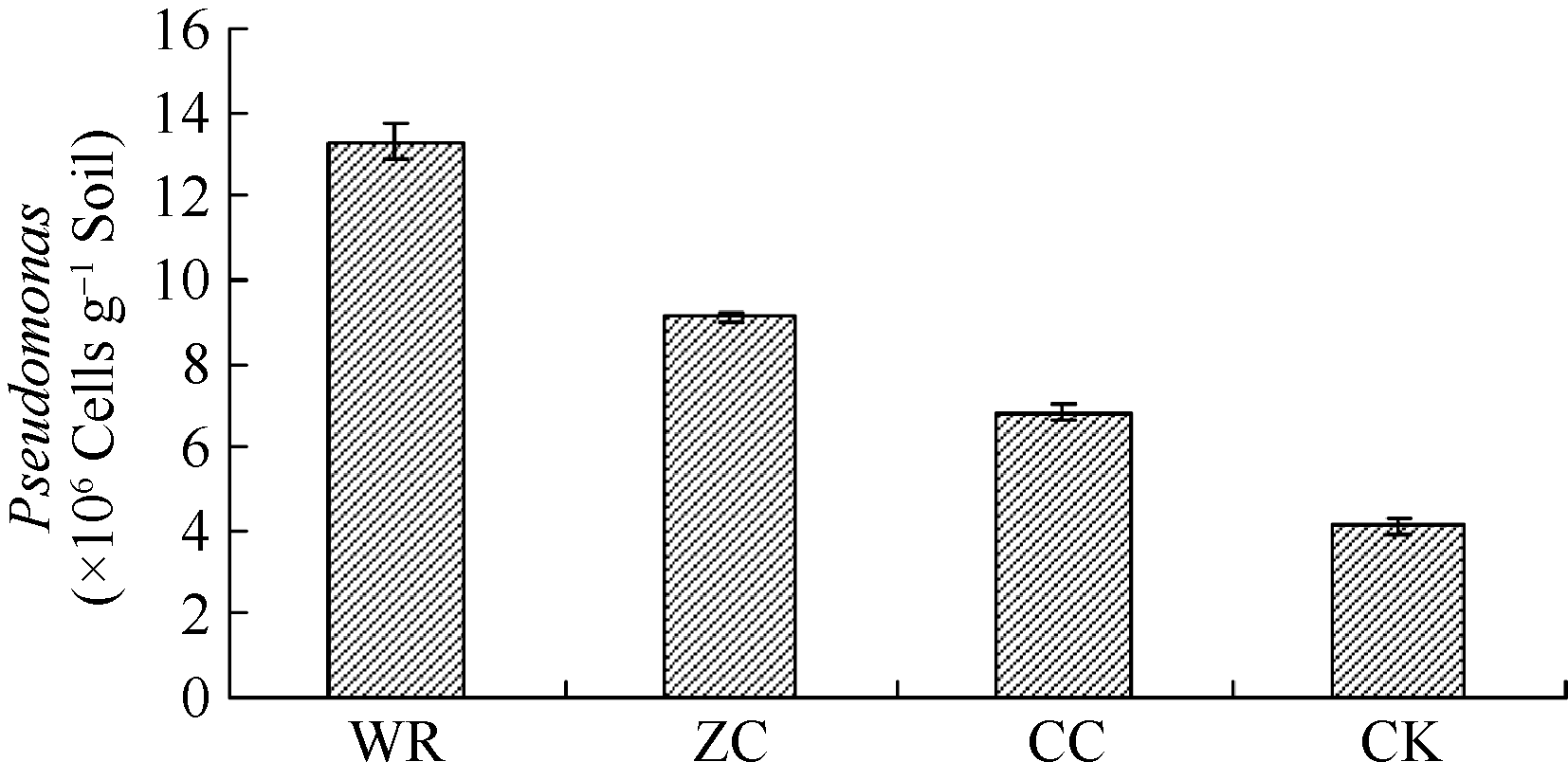 | 图7 不同连作年限地黄根际土壤假单胞菌属qRT-PCR定量分析CK: 对照土; ZC: 头茬土壤; WR: 野生土壤; CC: 重茬土壤。Fig. 7 qRT-PCR quantification ofPseudomonasin rhizospheric soils of R. glutinosa with different year monoculturesCK: control soil; ZC: newly planted soil; WR: wildR. glutinosa soil; CC: two-year monocultured soil. |
植物连作障碍形成及加重的原因极其复杂, 涉及土壤、植物、微生物等多个方面, 一些研究表明土壤理化性状的改变和土壤肥力缺乏是导致作物连作障碍发生的主要原因[24]。而本研究发现重茬土壤样品中碱解氮、速效磷、速效钾的含量都显著高于头茬和野生状态地黄根际土壤, 表明地黄连作并未导致土壤速效养分下降, 反而出现积累效应。王茂胜等[25]研究发现, 随着种植年限的增加, 植烟的品质和产量随之下降, 而土壤中全氮、全磷、有机质的含量逐渐增加。郭红伟等[26]也发现辣椒连作下, 土壤全磷、全钾、速效养分和大量元素都有不同程度的积累。鉴于此, 本研究进一步采用PLFA和T-RFLP 2种技术深入分析不同连作年限野生地黄以及野生状态地黄根际土壤微生物群落结构的变化。
运用PLFA法分析地黄根际土壤微生物量变化表明, 重茬地黄根际土壤总微生物量与头茬相比显著下降, 并且细菌/真菌比例下降。进一步, T-RFLP分析发现野生地黄连作导致土壤微生物群落结构逐渐发生偏移, 并且某些潜在病原菌(如Clostridium sp.、Flexibacter polymorphus和Clostridium ghoni)在重茬地黄根际土壤中大量滋生。不同连作年限处理下, 地黄根际土壤微生物群落结构存在较大差异可能与地黄根系分泌物介导有密切关系。有报道发现, 将黄瓜自毒物质— 香豆酸外源添加至土壤中, 会对土壤微生物群落结构产生显著影响, 而且导致厚壁菌门、β -变形菌门等细菌大量增加及某些病原菌大量繁殖增长[27]。同时, 本研究发现, 野生状态地黄和头茬地黄根际土壤中聚集了大量有益菌群, 如芽孢杆菌属(Bacillus)、类芽孢杆菌属(Paenibacillus)、假单胞菌属(Pseudomonas)等。芽孢杆菌属和假单胞菌属作为PGPR菌株(植物根际促生菌)已被广泛运用到农业生产中以提高作物的产、质量和防治病虫害[28]。尤其是假单胞菌属, 作为一种重要的生物防治菌株, 具有较好的生物防治效果, 能够有效抑制土传病害的发生[29, 30]。Jetiyanon等[31]报道, 通过混合使用PGPR菌株能够诱导植物产生系统抗性进而抵御多种植物病害。进一步, qRT-PCR绝对定量分析也证实了在野生状态地黄和头茬地黄根际土壤中假单胞菌的数量显著高于重茬根际土壤, 即野生地黄连作下根际微生态结构恶化, 有益菌含量下降, 导致连作植株易受病害侵染。分析还发现, Cytophaga在野生状态地黄和头茬地黄根际土壤中相对含量较高, Cytophaga对植物根系分泌物、纤维素、土壤有机质等降解有重要作用[32]。重茬地黄根际土壤中纤维素降解菌含量下降可能影响土壤微生态环境的营养循环, 从而降低地黄抵御病害的能力, 最终导致地黄产、质量下降。
与传统的分离培养方法相比, 新兴的分子生物学技术能够更加准确全面地反映微生物群落结构的组成和结构。本研究将PLFA与T-RFLP分析相结合, 系统分析不同连作年限地黄根际土壤微生物群落结构的差异, 并加以qRT-PCR定量验证, 结果更加全面准确。本研究结果对深入阐明药用植物连作障碍形成的分子机制及探索科学有效的缓解措施具有一定的理论与指导意义。
野生地黄在连作情况下仍然长势较差, 初步证实这与其根际土壤总微生物量下降, 细菌/真菌比例降低有一定相关性。野生地黄连作对其根际土壤细菌群落结构的组成及分布有很大影响, 有益菌数量较少, 病原菌大量滋生, 而野生状态地黄土壤和头茬地黄土壤中却聚集较多的有益生防菌, 以假单胞菌属、芽孢杆菌属最为显著, 不同栽培情况下野生地黄根际土壤假单胞菌属数量变化趋势显著。
The authors have declared that no competing interests exist.
作者已声明无竞争性利益关系。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|