
{kind=link}
{kind=link}
{kind=link}
{kind=link}
用全基因组关联分析解析籼稻垩白的遗传基础
[邱先进1, 2  , 袁志华
, 袁志华1 , 陈凯3 , 杜斌1 , 何文静1 , 杨隆维1, 2 , 徐建龙3, 4, * , 邢丹英1, 2, * , 吕文恺1 ]
, 袁志华, 邢丹英, 吕文恺|
|


第一作者联系方式: E-mail: xjqiu216@yangtzeu.edu.cn
利用272份全球籼稻微核心种质的重测序SNP基因型, 对海南三亚、广东深圳、浙江杭州和湖北荆州4个地点收集到的垩白粒率和垩白度性状采用TASSEL软件进行全基因组关联分析, 解析籼稻垩白的遗传基础和挖掘影响垩白粒率和垩白度的优异等位基因。结果表明, 依据SNP数据, 可将籼稻微核心种质分成3个亚群。4个地点分别检测到42个和44个与垩白粒率和垩白度显著关联的位点, 位于全部12条染色体上。2个性状分别有21个和19个位点在2个以上环境下同时被检测到, 这些位点中有12个位点同时影响垩白粒率和垩白度, 11个位点附近都有已克隆的水稻品质相关基因。其中, 第5染色体3.3~5.3 Mb区间在4个地点都被检测到与垩白粒率显著关联, 以杭州点对垩白粒率的贡献最大, 优异等位基因载体品种为IRGC121689; 第12染色体的17.5~18.0 Mb区间在三亚和杭州都被检测到与垩白度显著关联, 以三亚点的垩白度贡献最大, 优异等位基因载体品种为IRGC122285。这些位点和品种资源可作水稻外观品质分子改良的重要基因和品种资源。
, YUAN Zhi-Hua, XING Dan-Ying, LÜ Wen-KaiIn order to dissect the genetic bases and mine novel alleles of grain chalkiness in indica, we conducted an experiment with genome-wide association analysis using phenotypic data collected from multiple locations (Sanya of Hainan, Shenzhen of Guangdong, Hangzhou of Zhejiang, and Jingzhou of Hubei) and 6704 re-sequenced SNP markers distributed in whole genome for 272 indicamini-core germplasm collected worldwide. All accessions were classified into three subpopulations based on SNP data. Total of 42 and 44 loci were detected as significant associations with percentage of grains with chalkiness (PGWC) and degree of endosperm chalkiness (DEC), respectively, which distributed all over the 12 chromosomes. Twenty one and nineteen loci were stably expressed for PGWC and DEC in multiple locations, respectively, and 12 simultaneously affected the two traits. Eleven of the said loci were co-located in the same or near regions harboring the quality genes cloned previously. Of them, the region of 3.3-5.3 Mb on chromosome 5 was significantly associated with PGWC at all four locations, having the largest phenotypic contribution detected in Hangzhou location, and the carrier variety with the best favorable allele was IRGC121689; another region of 17.5-22.7 Mb on chromosome 12 was significantly associated with DEC at Sanya and Hangzhou, having the largest phenotypic contribution detected in Sanya, and the carrier variety with the best favorable allele was IRGC122285. These loci and germplasms are important potential genes and variety resources that can be used in molecular breeding for rice appearance quality.
水稻是世界上最重要的粮食作物之一。随着人们生活水平的提高, 水稻品质日益受到人们的重视。垩白是衡量水稻外观品质优劣的重要指标之一, 而且能通过影响整精米率影响水稻的加工品质[1, 2, 3]。因此, 阐述水稻垩白性状的遗传基础, 挖掘影响垩白性状的有利基因, 利用分子育种改良水稻外观品质, 培育品质优良的水稻新品种, 具有重要意义。
衡量水稻垩白的指标有垩白粒率和垩白度[4], 这2个性状都是典型的数量性状, 受多基因控制, 同时存在着明显的环境与基因型互作效应[5]。随着水稻基因组学的发展和分子标记技术的发展, 研究者利用各种群体定位了许多影响水稻垩白的QTL, 这些QTL分布于水稻的全部12条染色体上[6, 7, 8, 9, 10, 11]。其中qPGWC-7[12]和qPGWC-8[10]已经精细定位到了150 kb以下, GW2[13]和Chalk5[14]都已被成功克隆。此外, 利用水稻突变体结合图位克隆的方法也克隆了一大批影响水稻垩白的基因, 如OsPPDKB[15]、SSIII a[16, 17]、GIF1[18, 19]、ms-h[20]、fo-2[21]、OsRab5a[22]等。
目前大部分垩白QTL定位都是利用双亲作图结合图位克隆的方法获得的, 这种方法的群体构建时间长, 且只能鉴定出两个亲本中较好的等位基因, 无法获得种质资源中存在的最优等位基因, 而利用自然群体的关联分析正好可以弥补这一缺陷[23]。近年来, 利用全基因组关联分析定位水稻农艺性状的报道越来越多。Huang等[24]通过517份核心种质进行全基因组关联分析, 共定位了影响14个农艺性状的37个QTL, 其中一些位点与已知基因位于同一区间, 部分位点被直接精细定位到100 kb以内。随后, 他们又将群体扩大至950份, 对抽穗期和产量相关性状进行了全基因组关联分析, 在原有基础上定位了32个新位点[25]。Zhao等[26]利用来源于全世界不同地区的413份核心种质, 结合全基因关联分析的方法定位到影响34个性状的大量QTL。
本研究从国际水稻研究所引进296份世界籼稻微核心种质, 在海南三亚、广东深圳、浙江杭州和湖北荆州4个环境下考察分析了其中的272份能正常成熟的籼稻品种的垩白性状, 结合全基因组基因型数据解析籼稻垩白的遗传基础, 挖掘影响籼稻垩白的有利基因, 为分子育种改良水稻外观品质提供了基因和亲本资源, 为拓宽我国育种材料的遗传多样性奠定了基础。
272份籼稻微核心种质由菲律宾国际水稻研究所提供, 它们来自三大洲的31个国家, 其中非洲8个, 拉丁美洲13个和亚洲218个, 另有33个来源不明。
2012年冬季于海南三亚中国农业科学院作物科学研究所南繁基地, 2013年夏季于广东深圳中国农业科学院深圳基地、浙江杭州浙江省农业科学院基地和湖北荆州长江大学校内基地种植全部材料, 分别记录每个品种在4个地点的抽穗期, 272个品种的平均抽穗期在三亚为68.99 d, 在深圳为79.35 d, 在杭州为100.13 d, 在荆州为98.98 d。随后根据抽穗期将272个品种分组。2013年冬季和2014年夏季分别将这些材料按成熟期分组分期播种于上述4个环境, 保证272个品种的抽穗期基本一致, 最大限度地降低环境对垩白性状的影响。每个品种种植3行, 每行10株, 株行距为20 cm × 25 cm, 2次重复, 常规田间管理。为防止部分材料过高而倒伏, 抽穗后利用竹竿搭架。品种成熟后, 混收中间行的中间8株用于表型鉴定。
收获后自然晾干, 室温储藏3个月后参照国标GB/T 17891-1999[4]考察垩白性状。首先取30 g稻谷利用砻谷机和精米机将稻谷打成精米, 然后将全部精米利用JMWT12大米外观品质检测仪(东孚久恒, 北京)测定每个品种的垩白米粒, 重复测定2次。具有垩白的整精米粒数与所有整精米粒数的比值为垩白粒率(percentage of grains with chalkiness, PGWC), 垩白米粒中垩白面积与整个米粒面积的比值为垩白大小, 垩白粒率与垩白大小的乘积即为垩白度(degree of endosperm chalkiness, DEC)。对垩白粒率和垩白大小重复测定2次, 并计算垩白度, 取2次重复的平均值。
每个品种种植20株幼苗, 将幼嫩的叶片混合取样, 并参照Murry和Thompson提出的CTAB法[27]提取每个品种的DNA。利用GBS (genotyping by sequencing)技术获得SNP基因型, 由国际水稻研究所委托澳大利亚DArT公司完成[28]。基于原始获得的4万多个标记数据, 去掉无法比对到参考基因组或比对到全基因组存在多个位点的标记; 去掉插入/缺失标记, 保留SNP类型的标记; 去掉稀有等位基因(基因型频率< 5%)所在的标记; 去掉缺失标记数多于20%样本数的SNP标记。最终获得高质量的6704个有效SNP分子标记, 根据IRGSP1.0参考基因组确定所有标记的精确物理位置。
1.4.1 性状表型分析 用Microsoft Excel 2007整理数据, 计算各个性状在4个环境下的平均值、标准差、变幅、变异系数和广义遗传力, 并对4个地点的表型进行了方差分析。广义遗传力的计算参照盖钧镒等[29]介绍的方法。
1.4.2 遗传多样性分析 应用PowerMarker3.25软件[30]计算每个位点的等位基因数(allele number per locus)、遗传多样性(gene diversity)和多样性信息含量(polymorphism information content, PIC)。
1.4.3 群体结构分析 利用Structure 2.3.4软件[31, 32, 33, 34]对微核心种质进行群体结构分析, 确定亚群数, 并计算各品种归属于第k个亚群的概率Q值。在确定亚群时, 选用混合模型和独立等位基因频率模型依此设定亚群数为1~10, 将MCMC (Markov Chain Monto Carlo)开始时的迭代长度设置为50 000, 将其后的迭代次数设置为10 000, 每个K值独立运算5次。如果对数似然函数ln P(D)随着K值增大而增大, 则采用Δ K来确定合适的K值。Δ K为|L′ ′ (K)|的平均值除以L(K)的标准差, 其中L′ ′ (K) = L′ (K) - L′ (K+1), L′ (K) = L(K) - L(K-1), L(K)即为Structure 2.3.4运行结果中的ln P(D)。
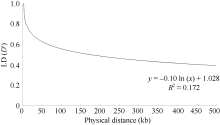
1.4.4 连锁不平衡(LD)分析 使用标准不平衡系数(D° )衡量位点之间的LD程度。D′ 值的变化范围为0~1, 将D′ 小于0.5作为LD衰减的标志。LD的计算过程在TASSEL5.0软件[35, 36]中完成。根据同一条染色体上标记间的物理距离和标记间的连锁不平衡程度, 通过回归分析计算LD随物理距离变化的回归方程y= aln (x)+b, 绘制LD衰减图, 用于观测LD与物理距离(kb)之间的关系。
1.4.5 垩白性状与SNP标记的关联分析 使用TASSEL5.0软件中GLM (general linear model)程序, 将Structure 2.3.4软件运行形成的Q值作为协变量, 然后利用标记变异分别对4个地点的垩白粒率和垩白度表型逐一进行回归分析。当P< 0.001时, 认为标记与性状关联显著, 此时R2值即为贡献率。
4个地点垩白粒率最小的为0, 最大的可达100%; 垩白度最小的为0, 最大的为86.3%, 表明籼稻微核心种质的垩白粒率和垩白度变异非常丰富。2个性状在4个地点表现十分一致(F值分别为0.5755和0.9708), 且广义遗传力较高(表1)。
| 表1 籼稻微核心种质在4个地点垩白性状的表现及广义遗传力 Table 1 Performance and heritability of rice chalkiness traits in indica mini-core germplasm across the four locations |
利用分布于全基因组的6704个SNP标记对全部272个品种进行遗传多样性分析, 共检测到14 356个等位基因, 大部分位点的等位基因为2个, 也有948个位点的等位基因为3个, 全基因组平均每位点2.1个等位基因。各位点的基因多样性平均为0.2977, 变幅为0.0501~0.6146。PIC平均值为0.2464, 变幅为0.0489~0.5336。
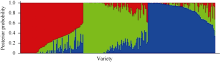
利用Structure 2.3.4软件对籼稻微核心种质进行群体结构发现, 对数似然函数[ln P(D)]随着亚群的增多而增大(图1-A), 无法确定最适宜的亚群数。然后采用Evanno等[37]介绍的方法进一步分析发现, 当K为3时Δ K最大(图1-B), 因而该群体最适宜的亚群数为3。再利用Structure 2.3.4计算的概率Q值将每个品种划分到相应的亚群中, 并将3个亚群命名为POP1、POP2和POP3 (图2)。6704个SNP标记中无论是否在同一条染色体上, 标记间都存在极显著的LD (P< 0.01)。D° 回归遵循方程y= -0.10 ln (x)+1.028 (R2=0.172)。当D° 取0.5时, LD衰减距离为196 kb, 适合做水稻的关联分析(图3)。
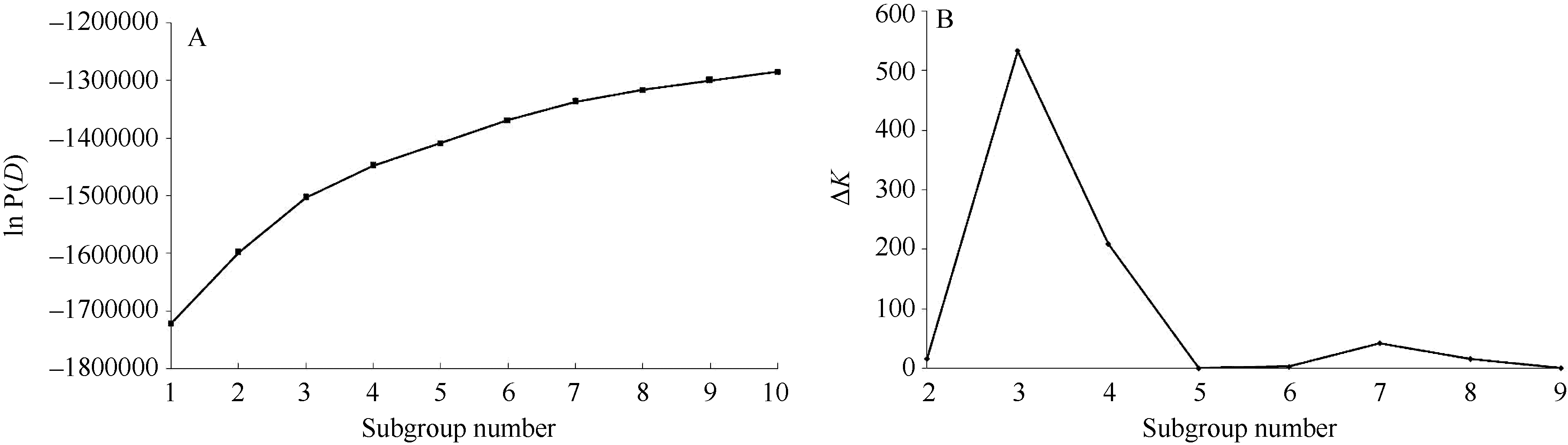 | 图1 籼稻微核心种质lnP(D)(A)和Δ K(B)随亚群数的变化Fig. 1 Change of ln P(D) (A) and Δ K (B) on the subgroup number in indica mini-core germplasm |
 | 图2 利用Strucute 2.3.4计算的籼稻微核心种质中每个品种归属于3个亚群的后验概率Fig. 2 Posterior probability of each rice variety of indica mini-core germplasm belonging to the three subpopulations calculated by Structure 2.3.4 software |
 | 图3 共线性SNP标记位点之间的距离与D° 之间的关系Fig. 3 Relationship between D° -value and physical distance of syntenic SNP markers |
利用PowerMarker 2.35计算了Nei’ s遗传距离显示, 3个亚群的遗传距离都较小, 其中POP1和POP3的遗传距离最大(0.0518), POP2和POP3的遗传距离最小(0.0399)。3个亚群的遗传多样性, 以POP1较高, 基因多样性指数为0.2751, PIC值为0.2267 (表3), POP3最小, 基因多样性指数为0.2528, PIC值为0.2088。相比较而言, 每个亚群的遗传多样性都比总体的遗传多样性低, 说明各个亚群在分化中都有部分等位基因被固定下来。
| 表2 3个亚群的基因多样性和多态性信息含量 Table 2 Gene diversity and polymorphism information content (PIC) value for three subpopulations |
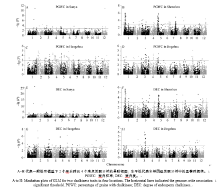
在海南三亚、广东深圳、浙江杭州和湖北荆州分别检测到15、14、24和16个位点与垩白粒率显著关联(图4)。2个或2个环境以上均检测到的与垩白粒率显著关联的位点有21个 (表3), 在12条染色体上均有分布, 贡献率为4.98%~15.80%。贡献率最大的是位于第5染色体的3.3~5.3 Mb区间, 在4个地点都检测到该区间与垩白粒率关联显著, 且在杭州贡献率最大, 达到15.8%, 不同等位基因间表型差异达到22.56% (表4), 有利等位基因载体品种是IRGC121689; 该位点在深圳对垩白粒率的贡献率为15.11%, 不同等位基因间表型差异达到22.31%, 有利等位基因载体品种为IRGC121858; 这个区间里含有1个已克隆的垩白基因Chalk5。贡献率最小的是位于第9染色体5.1~6.3 Mb区间, 在三亚的贡献率最小, 为4.98%。
对垩白粒率贡献率前5位的位点等位基因间表型差异都达到了15%以上, 其中等位基因垩白粒率差异最大的是第11染色体24.2~25.0 Mb区间。在深圳和杭州该位点都检测到与垩白粒率显著关联, 其中在杭州不同等位基因间垩白粒率差异高达31.89%, 有利等位基因载体品种是IRGC122290。在三亚、杭州和荆州, 第4染色体0.8~1.0 Mb区间都与垩白粒率显著关联, 其中在杭州对垩白粒率的贡献率达到14.32%, 不同等位基因间垩白粒率差异为28.98%, 有利等位基因载体品种是IRGC117623。在深圳和杭州, 第1染色体5.8~7.8 Mb区间都与垩白粒率显著关联, 其中在深圳对垩白粒率的贡献率达到11.42%, 不同等位基因间垩白粒率差异为16.86%, 有利等位基因载体品种是IRGC117531。
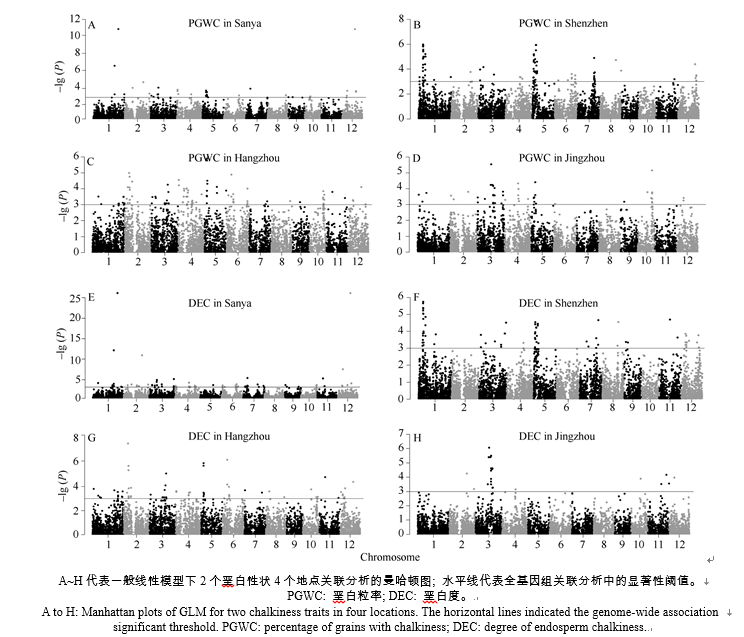 | 图4 籼稻微核心种质4个地点垩白性状全基因组关联分析Fig. 4 Genome-wide association analysis of chalkiness traits in four locations using indica mini-core germplasm |
| 表3 全基因组关联分析检测到不同环境垩白性状的显著关联位点 Table 3 Significant loci associated with the two chalky traits detected by genome-wide association analysis |
| 表4 与垩白性状显著关联且贡献率位于前5位的位点上等位基因效应差值及带有优异等位基因的载体品种 Table 4 Allelic differences for the top five loci associated with chalkiness traits and their typical carrier variety with the best favorable alleles |
在4个地点分别检测到24、14、23和7个位点与垩白度显著关联(图4)。2个或2个环境以上均检测到的与垩白度显著关联的位点有19个 (表3), 分布于除第8染色体外的所有染色体, 贡献率为5.60%~45.43%。贡献率最大的是位于第12染色体的17.5~18.0 Mb区间, 在三亚和杭州均检测到该区间与垩白度显著关联, 其中在三亚贡献率高达45.43%, 不同等位基因间垩白度差异达到14.44%, 有利等位基因载体品种是IRGC122285; 贡献率最小的是位于第9染色体1.2~2.5 Mb区间, 在深圳的贡献率最小, 为5.6%。
对垩白度贡献率前5位的位点不同等位基因间垩白度差异有很大差别(表4), 最大的是第2染色体5.5~6.1 Mb区间。在三亚和杭州试点, 该位点都与垩白度显著关联, 其中在杭州对垩白度的贡献率为19.26%, 不同等位基因间垩白度差异高达16.29%, 有利等位基因载体品种为IRGC117271。在深圳和杭州试点, 第5染色体3.3~3.4 Mb区间都与垩白度显著关联, 其中在杭州对垩白度的贡献率达到15.15%, 不同等位基因间垩白度差异为10.34%, 有利等位基因载体品种是IRGC121855。在深圳和杭州试点, 第2染色体24.0~26.0 Mb区间都与垩白度显著关联, 其中在杭州对垩白度的贡献率达到22.24%, 不同等位基因间垩白度差异为7.03%, 有利等位基因载体品种是IRGC121853。在三亚、深圳和荆州试点, 第12染色体5.8~7.1 Mb区间都与垩白度显著关联, 其中在三亚对垩白度的贡献率达到15.67%, 不同等位基因间垩白度差异为5.68%, 有利等位基因载体品种是IRGC121855。
本研究利用6704个SNP标记对272份籼稻微核心种质进行基因型分析, 86%的标记位点上的等位基因有2个, 全基因组每个标记位点的平均等位基因数为2.14个, 平均基因多样性为0.30, PIC平均多态性信息(PIC)为0.25, 说明本研究所用的籼稻微核心种质群体的遗传多样性比较丰富。但本研究中, 各项遗传多样性参数小于前人的研究结果[44]。究其原因, 主要是本研究的材料是从全球种质资源中筛选得到的籼稻微核心种质, 是水稻的一个亚种, 筛选过程中人为降低了样本的遗传多样性。另外, 本研究鉴定基因型所用的标记为SNP标记, 大部分都只能鉴定2种或3种等位基因, 比SSR标记等其他标记检测的等位基因数少。
对关联分析影响较大的几个因素包括LD大小、交配类型、群体大小和群体结构等, 其中群体结构和LD对关联分析的影响非常大。群体结构主要影响到关联分析的准确性。到目前为止, 已经有许多学者对水稻的群体结构进行了研究。他们利用SSR标记将种质群体分为5个或更多个亚群。Zhao等[26]利用覆盖全基因组的44 100个SNP将来源于82个国家的413份水稻品种分为5个亚群; Huang等[24]利用二代测序将517个水稻品种分为籼稻和粳稻两个亚群。虽然这些分群结果很不一致, 但都说明水稻存在普遍的群体结构。然而, 对于水稻亚种的再分群研究较少。Huang等[24]将517个水稻品种分成籼稻和粳稻后, 又在亚种内进行了群体结构分析, 结果籼稻和粳稻都可分为3个亚群。本研究利用6704个SNP标记将272份籼稻微核心种质分成了3个亚群, 与Huang等[24]的结果类似。LD主要影响关联分析的精确性。Zhao等[26]认为籼稻的LD距离约为100 kb, Huang等认为是123 kb。本研究通过6704个SNP标记研究的结果表明籼稻的LD距离为196 kb, 与他们的研究结果差异不大。
本研究在海南三亚、广东深圳、浙江杭州和湖北荆州分别检测到42个和44个位点与垩白粒率和垩白度显著关联, 其中在2个以上地点同时检测到的与垩白粒率和垩白度显著关联的位点分别是21个和19个。这些位点中, 有11个位点区间内或附近含有已克隆的或精细定位的水稻品质基因, 说明本研究的结果具有较高的可靠性。这11个位点根据附近基因的类别可分为3类, 第一类为垩白基因, 如GW2、GIF1、Flo2、Chalk5、qPGWC-7和OsRab5a; 第二类为水稻淀粉含量基因Wx; 第三类为水稻粒型和粒重基因, 如GS2、GS3、TGW6和GW8。直链淀粉含量基因、粒型基因和粒重基因都与垩白性状有显著关联, 究其原因, 垩白是在灌浆过程中发生的一种现象, 前人研究发现垩白与淀粉合成与运输的各个途径相关, Wx是淀粉合成过程中的关键基因, 因此与垩白显著关联; 在灌浆过程中, 淀粉运输是从背部到腹部, 籽粒腹部因为运输路途长而易产生垩白[45]。因此垩白与粒型具有显著的负相关。而决定粒重的主要是粒型, 因此粒型基因和粒重基因位点都可能与垩白性状显著关联。
在2个以上环境下检测到与垩白性状显著关联的位点中, 有12个位点同时与垩白粒率和垩白度显著关联, 占全部垩白位点的60.0% (24/40), 说明垩白粒率与垩白度的遗传基础高度一致。这12个位点中含有已知品质基因的有6个, 另有6个位点是没有克隆的垩白相关基因, 分别是第1染色体5.8~7.8 Mb、第3染色体0.7~4.0 Mb、第4染色体0.8~1.0 Mb、第10染色体18.8~22.3 Mb、第11染色体24.2~26.6 Mb和第12染色体5.8~7.1 Mb。对于这些位点垩白相关基因的精细定位和克隆, 将进一步丰富对垩白性状的分子调控机制的认识, 从而为改良水稻垩白提供新基因。6个已有克隆基因的位点中, 2个位点都与粒型基因(GS2和GS3)有关, 这些基因是否直接控制垩白或其与垩白基因存在紧密连锁, 还需要后续验证。
垩白的遗传基础十分复杂, 受环境影响大。利用分子标记辅助选择改良水稻垩白是一种十分高效的方法。本研究关联到的位点中, GS2、GS3、GIF1、Chalk5、qPGWC-7、第3染色体0.7~4.0 Mb、第4染色体0.8~1.0 Mb和第12染色体5.8~7.1 Mb区间这些个位点在3个或4个地点都与垩白粒率和垩白度显著关联, 可作为分子育种改良水稻垩白的重点基因位点或区域。其中GS2、Chalk5、第4染色体0.8~1.0 Mb和第12染色体5.8~7.1 Mb区间对垩白粒率和垩白度的贡献率较大, 携带这些位点优异等位基因的品种有IRGC121689、IRGC121858、IRGC 117623、IRGC121853、IRGC122025和IRGC121855, 可优先利用。值得注意的是, 在利用这些位点改良水稻垩白性状要同时考虑它们可能产生的负向效应。例如, 本研究中Chalk5在4个环境下均与垩白显著关联, 且对垩白的贡献率很高, 但是这个位点与控制粒宽的基因GS5[46]紧密连锁, 两者相距104 kb, 在改良垩白的同时会造成粒宽变小从而降低粒重, 最终影响产量。因此, 要打破这种不利连锁累赘, 必须种植非常大的群体。或将一些只影响垩白而与粒形无关的稳定表达基因qPGWC-7与位于第3染色体5.7~10.8 Mb、第4染色体0.8~1.0 Mb和第12染色体5.8~7.1 Mb区间的主效基因进行标记辅助聚合, 可以在不损失产量的前提下实现外观品质的改良。
272份籼稻微核心种质可分为3个亚群, 其连锁不平衡衰减距离为196 kb。在海南三亚、广东深圳、浙江杭州和湖北荆州4个环境分别检测到与垩白粒率和垩白度显著关联的42个和44个位点, 其中21个和19个位点在2个以上环境下都被检测到, 12个位点同时影响垩白粒率和垩白度, 11个位点附近已克隆了品质相关基因。GS2、GS3、GIF1、Chalk5、qPGWC-7、第3染色体5.7~10.8 Mb、第4染色体0.8~1.0 Mb和第12染色体5.8~7.1 Mb区间这些个位点在3或4个地点都与垩白粒率和垩白度显著关联, 其中GS2、Chalk5、第4染色体0.8~1.0 Mb和第12染色体5.8~7.1 Mb区间对垩白粒率和垩白度的贡献率最大, 携带这些位点优异等位基因的品种有IRGC121689、IRGC121858、IRGC117623、IRGC121853、IRGC122025和IRGC121855, 这些位点和品种可用作垩白性状分子改良的重要基因和品种资源。
The authors have declared that no competing interests exist.
作者已声明无竞争性利益关系。The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|