{kind=link}
{kind=link}
利用简化基因组技术分析甘薯种间单核苷酸多态性
[石璇1 , 王茹媛1 , 唐君2 , 李宗芸1, *  , 罗永海
, 罗永海1, * ]
, 罗永海]
|
|


第一作者联系方式: E-mail: shixuanluck@163.com
选用Xushu 18 (6 x)和Nancy Hall (6 x)、2个二倍体 I. trifida (2 x)品系(4597-10和4597-21)、四倍体 I. trifida (4 x)、六倍体 I. trifida (6 x)、二倍体 I. temussima (2 x)和 I. littorallis (2 x)以简化基因组测序技术SLAF-seq测序, 获得724 589个SLAF标签, 其中多态性SLAF标签35 310个。通过序列分析, 获得40 765个有效单核苷酸多态(SNP), 并用这些SNP分析了8个种质的群体结构和系统发生树。结果表明, 利用简化基因组测序技术SLAF-seq能高效、低成本地开发出大量可用于群体遗传分析的SNP标记; 通过构建进化树发现甘薯栽培种和野生种 I. trifida的亲缘关系比较近。这些分析结果为进一步研究甘薯栽培种的起源提供了基础数据。
Xushu 18 (6 x), Nancy Hall (6 x), I. trifida (2 x) 4597-10, I. trifida (2 x) 4597-21, I. trifida (4 x), I. trifida (6x), I. temussima (2 x), and I. littorallis (2 x) were used as experimental materials for sequencing by specific-locus amplified fragment sequencing (SLAF-seq), a high-throughput reduced-representation genotyping technology. In total, 724 589 SLAF tags were obtained and 40 765 SNPs were identified out of 35 310 polymorphic SLAF tags. A total of 40 765 single nucleotide polymorphisms (SNPs) were obtained by sequence analysis. Population structure and phylogenetic relationship of eight germplasm were analyzed using the SNP dataset, which suggests that SLAF-seq can be used to develop large-scale SNPs for population genetic analysis, effectively and economically. Our analysis revealed that the relationship between sweet potato cultivars and the wild species I. trifida is closer . These results provide empirical data for further study of the origin of sweet potato.
甘薯[Ipomoea batatas (L.) Lam]又称甜薯、地瓜、白薯、红薯, 旋花科甘薯属草本植物, 是世界范围内最重要的经济作物之一[1]。甘薯是六倍体作物(2n = 6x = 90), 由于形态相似程度高、进化过程中发生难以追踪的杂交以及表型可塑性等问题, 很难仅仅根据甘薯及其近缘物种的形态学特征来构建其系统发生树[2, 3]。时至今日, 甘薯的起源和驯化历史仍不明确。现有的研究表明, 甘薯有可能起源于异源多倍体, 其祖先种可能是I. leucantha、I. triloba和I. trifida[4]; 但是更多的研究提示甘薯可能起源于同源多倍体, 推测由二倍体I. trifida染色体加倍而成[2, 3, 5, 6, 7, 8]。尽管如此, 现有数据并不足以确定甘薯的起源关系, 特别是缺少高质量的I. trifida和I. batatas的核基因组序列和叶绿体基因组序列, 不能通过比较基因组学分析证明二倍体的I. trifida是否是六倍体的甘薯栽培种I. batatas的祖先。
随着现代生物技术的发展, 分子标记越来越广泛地被用于确定物种间的亲缘关系。早期人们利用RAPD、RFLP、AFLP、ISSR等分子标记技术来构建甘薯遗传图谱, 但由于准确性差、工作量大、可重复性差、成本高等问题[2, 9, 10, 11, 12], 都无法开发出高通量的分子标记。随着测序技术的发展以及因此带来的测序成本降低, 高通量测序技术越来越多地被应用于分析复杂物种间的亲缘关系。在过去几十年, 基因组学蓬勃发展, 众多高等植物的基因组如拟南芥、水稻、苜蓿草、大豆、小麦等均已经完成了测序, 但是甘薯基因组的测序仍未成功。甘薯基因组构成复杂, 异质性高, 缺乏高解析度的遗传图谱。即便在今天的技术条件下, 要克服这些障碍、对甘薯进行全基因组测序和组装依然困难重重。此外, 甘薯是一种异花授粉的作物, 一般情况下不仅品种内自交不亲和, 同一种群内品种间也存在自交不亲和现象。这些情况严重限制了甘薯遗传学、基因组学和育种学的研究, 提示研究人员应该从研究方法或技术手段上多些创新和尝试。
简化基因组测序是在高通量测序技术(主要是二代测序技术)基础上发展起来的, 它利用酶切技术、芯片捕获技术或其他实验手段降低物种基因组复杂程度, 进而研究基因组代表性遗传变异的技术手段[13, 14]。SLAF-seq (specific-locus amplified fragment sequencing)是特异性位点扩增片段测序技术的简称, 是基于高通量测序技术发展起来的一种简化基因组深度测序技术。简要地说, SLAF-seq先利用生物信息学方法, 对目标物种的参考基因组(例如目标物种已知的BAC序列或近缘物种的基因组)系统分析; 再根据参考基因组的GC含量、重复序列情况和基因特点等信息, 设计酶切方案, 构建SLAF-seq文库, 筛选特异性长度片段, 应用高通量测序技术获得高通量标签序列; 然后对数据分析, 获取测序深度和质量均满足要求的SLAF片段; 这些片段可以充分代表全基因组的序列特征信息, 依据这些片段可以开发出大量的分子标记特别是单核苷酸多态(SNP)。该技术可以且已经运用于大规模种质资源研究、高密度遗传连锁图谱构建、不同个体间的多态性分析和目标性状相关的基因组区段或候选功能基因快速定位。它具有高准确性、高通量、低成本、获得的分子标记能作为序列信息直接进行后续工作、能缩短试验周期、可以多性状并行研究等优点[15, 16, 17]。本文尝试将SLAF-seq技术应用于甘薯生物学的研究, 测定8个甘薯栽培种及其近缘物种基因序列, 拟揭示品种间亲缘关系, 有效提高在复杂农作物中开发大规模分子标记、研究其物种起源与进化关系的能力。
选用8个甘薯或其近缘物种, 分别是六倍体栽培种Xushu 18 (6x)和Nancy Hall (6x)、2个二倍体I. trifida (2x)品系(4597-10和4597-21)、四倍体I. trifida (4x)、六倍体I. trifida (6x)、二倍体I. temussima (2x)和I. littorallis (2x)(基本信息列于表1)。
| 表1 本研究所用的8种甘薯或其近缘品种 Table 1 Eight germplasms of sweetpotato and its relatives in this study |
用液氮速冻新鲜叶片, 存于-80℃冰箱备用, 用CTAB法分别提取8个样品的基因组DNA, 用Nanodrop 2000C和Qubit 2.0进行DNA的质量检测, 以确保所提基因组DNA质量达到建库要求(即OD260与OD280的比值分布在1.8~2.0之间; DNA浓度达到30 ng μ L-1)。
由于没有合适的甘薯或其近缘物种参考基因组可用, 我们选择马铃薯基因组为参考基因组进行电子酶切预测。马铃薯基因组大小约为651 Mb, GC含量为34.80%[18]。利用SLAF酶切预测软件对参考基因组进行酶切预测, 选择最合适的酶切方案, 其原则如下: (1)位于重复序列的酶切片段比例尽可能低; (2)酶切片段在基因组上尽量均匀分布; (3)酶切片段长度与具体实验体系的吻合程度较高[17]; (4)最终获得酶切片段(SLAF标签)数达到预期标签数。同时, 通过设置对照基因组(拟南芥)进行同样流程的测序、评估对照基因组的测序数据, 进而验证实验过程是否正常, 确定所用酶切方案的有效性。
根据选定的最适酶切方案, 对各样品基因组DNA分别进行酶切, 分离目标酶切片段, 对其进行3° 端加A处理、连接测序接头、PCR扩增、纯化、混样、纯化等试验操作, 文库质检合格后用Illumina HiSeq 2500进行双端测序(PE100)。对测序得到的原始数据进行识别、过滤、质检、评估等分析, 获取各个样品的序列(reads)。
测序产生的序列(reads)来源于同一限制性内切酶对不同样品作用产生的长度相同或相近的酶切片段, 根据相似性可以将各样品的序列(reads)聚类, 聚到一起的序列(reads)被认为来源于同一个SLAF酶切片段, 称为一个序列组(SLAF标签)。一般来说, 同一SLAF标签在不同样品间的序列相似度远高于不同SLAF标签间的相似度; 同一个SLAF标签在不同样品间序列有差异, 即可定义为多态性SLAF标签。通过分析多态性SLAF标签, 鉴定其中存在的单核苷酸多态性(SNP), 利用这些SNP分析群体。
利用SLAF酶切预测软件分析, 最终确定使用Rsa I酶切, 酶切片段长度在264~314 bp的片段定义为目标酶切片段, 预测能得到107 700个SLAF标签。试验中, 从对照基因组(拟南芥)获得了1.79百万序列(MReads)。通过序列分析可知, Rsa I的酶切效率为93.50%, 双端比对效率(一条序列两端在对照基因组上的比对跨度介于50~1000 bp的reads占总reads的比例)为85.15%。这些数据显示我们选定的酶切方案和试验结果均达到试验预期。从8个甘薯栽培种及其近缘物种共获得18.66百万序列(MReads), 平均Q30 (测序碱基质量值, 即测序时错误识别的概率是0.1%、正确率为99.9%的碱基比例)为90.74%, 平均GC含量为38.69% (表2)。所测序列的GC含量处于较低水平, 对本试验测序反应的影响不大; Q30数据表明测序质量很高, 测序结果可靠。
| 表2 SLAF-seq基因组测序数据统计表 Table 2 Summary of genomic sequences generated by SLAF-seq |
通过序列分析, 从8个甘薯甘薯栽培种及其近缘物种共获得了724 589个SLAF标签, 每个样品获得的SLAF标签数从123 205至292 800不等(表3)。每个样品的平均测序深度(即各个样品中平均每个SLAF标签上对应的测序reads数)有所差别, 从6.62至17.03, 平均测序深度为11.46, 达到了SLAF试验预期(一般认为, 测序深度达到5即可提供较可靠的、高通量的分子标记, 可用于研究物种的亲缘关系)。
| 表3 SLAF标签统计表 Table 3 Summary of obtained SLAF tags |
从这些SLAF标签中共鉴定到多态性SLAF标签35 310个。通过分析, 鉴定到89 375个SNP (表4): 每个样品的SNP数目从7 096至14 860不等。这些SNP的平均完整度(即SNP所在位点在全部样品中的信息完整度)介于17.41%~36.45%之间。根据次要基因型频率(MAF)大于5%和完整度大于80%的选择标准, 从这些SNP中筛选出40 765个有效SNP, 用于群体遗传分析。
| 表4 群体SNP统计表 Table 4 Summary of identified SNPs |
此外, 分析所鉴定到的SNP可以看到, 不同样品的SNP杂合率存在较大差异, 多数样品具有较高的杂合率。其中六倍体栽培种Xushu 18 (6x)和Nancy Hall (6x)、野生种四倍体I. trifida (4x)、六倍体I. trifida (6x)的SNP杂合率都超过了92% (表4), 表明它们的基因组高度杂合。相反地, 野生种I. temussima (2x)和I. littorallis (2x)的SNP杂合率分别只有7.61%和15.05%, 表明其基因组的纯合度很高(表4)。
基于筛选出的有效SNP, 采用Admixture软件计算了8个甘薯栽培种及其近缘物种的群体结构(图1)。根据交叉验证错误率来确定分群数, 拥有最低交叉验证错误率的分群数为最优分群数。从图1可见, 当K为8时值最低, 这反映了8个甘薯栽培种及其近缘物种的遗传结构并不明显。
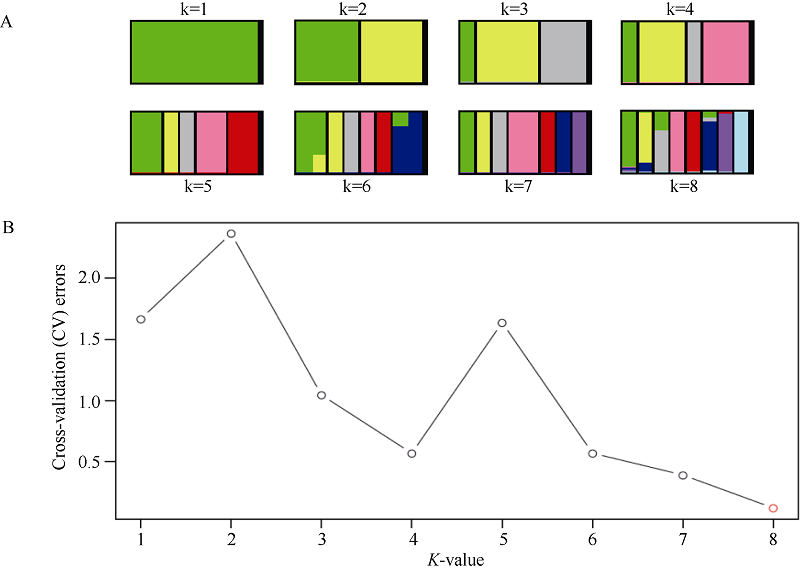 | 图1 基于所鉴定SNP分析群体结构A: 不同分群数(K值)的聚类结果显示无明显群体结构存在; B: 不同K值对应的交叉验证错误率显示k为8的时候交叉验证错误率最小。Fig. 1 Analysis of population structure by using identified SNPsA: cluster results of different structuring number (K-values) show no obvious population structure; B: cross validation error rates corresponding to different K-values show k = 8 is the best. |
利用这些SNP信息, 绘制8个甘薯栽培种及其近缘物种的进化树(图2)。从进化树可见, 2个六倍体栽培种Xushu 18 (6x)和Nancy Hall (6x)的亲缘关系最近; 二倍体I. trifida (2x)的2个品系在进化树上相邻, 显示它们更高的亲缘关系。值得关注的是, 2个六倍体栽培种与四倍体I. trifida (4x)和六倍体I. trifida (6x)比二倍体I. trifida (2x)的亲缘关系近。此外, 可看出, I. temussima (2x)和I. littorallis (2x)与甘薯栽培种的亲缘关系较远。
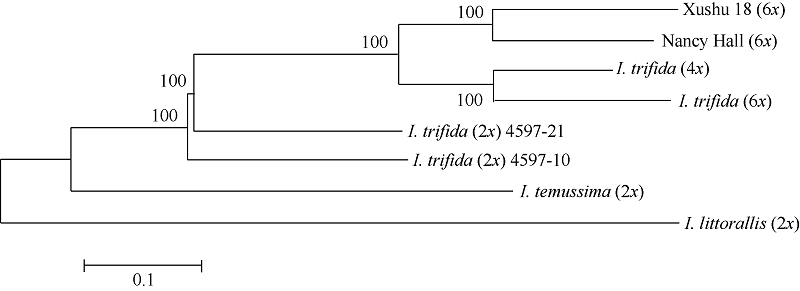 | 图2 基于所鉴定SNP的进化树Fig. 2 Phylogenetic tree based on identified SNPs |
分子标记技术是生物学研究不可或缺的技术手段。尽管我们已经步入后基因组时代, 在很多物种中, 分子标记依然十分有限。甘薯栽培种染色体数目多(2n = 6x = 90), 遗传背景复杂, 严重影响甘薯分子生物学和分子育种的研究[19]。早期人们利用RFLP来研究甘薯及其近缘野生种的亲缘关系, 但是花费较高, 工作量较大, 效率相对较低。也有研究者使用RAPD技术来研究种质资源间的遗传多样性, 但其结果的准确性及可重复性的问题很难解决[2]。近年来, Jarret等[3, 9]和贺学勤等[10]综合运用RAPD、AFLP和ISSR等分子标记分析并明确了一些甘薯品种的亲缘关系, 发现ISSR标记产生的聚类图与系谱图最吻合, 因而认为用ISSR标记分析甘薯品种的亲缘关系具有较高的效率和准确度。但是, ISSR标记存在一些局限性, 限制了其进一步的发展与应用。这些局限性包括, 需要一定的时间来摸索反应条件、筛选标记; 系显性遗传标记, 不能区分显性纯合基因型和杂合基因型; 通量小、精确性较低、耗时耗力、成本高等[11, 12]。尽管人们已经利用这些传统的分子标记技术研究了甘薯的亲缘关系、构建了一些甘薯及其亲缘物种的遗传图谱[23, 24, 25, 26, 27, 28, 29], 但是利用高通量测序技术开发全基因组范围内的分子标记并用于亲缘关系分析的报道仍然缺少。本文尝试应用简化基因组技术之一的SLAF-seq在甘薯及其近缘物种间开发大规模SNP, 并取得了成功。
简化基因组测序对没有参考基因组的物种特别适用, 能有效克服基因组复杂、序列与标记信息缺乏等问题, 进而大规模的开发和分析SNP标记。目前主要发展起来的简化基因组测序技术有, 限制性酶切位点相关的DNA(RAD)测序、基因分型测序(GBS)、基于II B型限制性内切酶位点相关的DNA (2b-RAD)测序和特异性长度扩增片段测序(SLAF-seq)技术。这些技术都是通过限制性内切酶来降低基因组DNA的复杂程度[30]。由于酶切产生的标记是全基因组范围内呈现特异性酶切位点附近的小片段DNA, 它们一定程度上代表了整个基因组的序列特征, 因此对酶切标签进行二代测序能够在大多数物种中获得成千上万的单核苷酸多态性(SNP)标记。本研究运用SLAF-seq技术, 在8个甘薯栽培种或其近缘物种中共获得18.66百万序列(MReads), 开发了724 589个SLAF标签(其中多态性SLAF标签35 310个), 从中鉴定到89 375个SNP, 进一步分析筛选出40 765个有效SNP并利用这些分子标记分析了相应种质的杂合率和群体结构, 构建了它们的系统发生树, 从而为甘薯栽培种的起源提供了新的证据。我们的研究表明, 利用简化基因组测序技术SLAF-seq能高效、低成本地开发出大量可用于群体遗传分析的SNP标记(表4)。这为下一步利用该技术在更大范围内分析甘薯栽培种及其近缘种的亲缘关系, 为阐明甘薯栽培种的起源历程创造了可能性, 同时也为开发分子标记用于基因定位(例如, 全基因组关联分析)和分子育种奠定了基础。
针对甘薯的起源目前主要有3种假说[31]。第一种假说认为甘薯起源于二倍体的I. leucantha, 然后通过染色体多倍化派生出四倍体的I. littoralis[32], 这2个物种再杂交产生三倍体的I. trifida, 进而染色体加倍产生六倍体的野生甘薯, 对这些野生甘薯进一步的筛选和驯化即产生了六倍体的I. batatas。第二种假说认为甘薯的野生祖先是I. batatas, 它是由I. trifida和I. triloba杂交产生的六倍体[33]。第三种假说认为I. trifida的同源多倍体是甘薯的祖先, 有可能是其产生的2n配子参与了一系列的染色体多倍体化[33]。此外, Roullier等[12]、Caroline等[34]、Otto[35]、Zohary[36]和Allaby等[37]通过分析ITS序列和SSR分子标记, 推测栽培种甘薯是从I. trifida进化而来的同源六倍体。他们发现, I. triloba与甘薯栽培种的亲缘关系比I. trifada更远, 无法得出其与甘薯存在明显的亲缘关系, 也就无法确切地证明甘薯是来源于I. trifida和I. triloba的异源六倍体[35]。从我们的数据可以看出, 甘薯栽培种和野生种I. trifida的亲缘关系比较近, 而且, 2个六倍体甘薯栽培种与四倍体I. trifida (4x)和六倍体I. trifida (6x)的比二倍体I. trifida (2x)的亲缘关系更近(图2)。这个数据与之前甘薯栽培种可能起源于I. trifida同源多倍化的假设相一致, 提示了I. trifida (2x)可能先多倍化形成I. trifida (4x), 之后进一步形成I. trifida (6x), 再经自然选择或者人工驯化形成甘薯栽培种[11, 23, 31, 34]。当然, 该假说需要进一步的基于比较基因组学、群体遗传学的分析以便获得无可辩驳的证据。
通过简化基因组测序技术SLAF-seq在甘薯栽培种及其近缘野生种中成功开发了高通量SNPs, 并将其用于亲缘关系的分析, 为进一步研究甘薯的起源提供了基础数据。
The authors have declared that no competing interests exist.
作者已声明无竞争性利益关系。The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|