



{kind=link}
{kind=link}
{kind=link}
主要麻类作物的ITS序列分析与系统进化
[张力岚1, 2 , 王俊1 , 万雪贝1, 2 , 徐益1, 2 , 张列梅1 , 方平平1 , 祁建民1, *  , 张立武
, 张立武1, 2, * ]
, 张立武]
|
|


第一作者联系方式: E-mail:1204549467@qq.com
比较主要麻类作物和测序植物间的ITS序列, 可明确它们间系统位置和进化关系。本研究采用PCR扩增和搜索GenBank数据库, 获得32份麻类作物和11份测序作物的核糖体内转录间隔区(internal transcribed spacers, ITS)序列, 利用MEGE软件分析ITS长度、G+C含量与同源性百分比差异。结果表明, 黄麻属、红麻属、苎麻属和亚麻属的ITS基本序列全长分别为963、939、658和686 bp; G+C含量分别为57.87%、58.03%、59.05%和53.75%。黄麻属变异区域集中在220~386 bp间, 红麻属变异区域集中在2个区段(206~347 bp, 599~713 bp), 苎麻属ITS变异区域分布在4个区段(158~163 bp、193~199 bp、288~333 bp和681~688 bp), 亚麻属ITS变异区域分布在5个区段(219~229 bp、235~240 bp、427~432 bp、468~484 bp和588~594 bp)。系统位置分析表明, 红麻属与棉花亲缘关系最近, 黄麻与棉花亲缘关系较近; 亚麻与苎麻各为一小支。系统位置分析与传统的植物分类结果较一致。研究主要麻类作物比较基因组学时, 红麻、黄麻可参考棉花, 苎麻可参考杨树或蓖麻。推测红麻属的进化时间约为33.7百万年前(million years ago, MYA), 黄麻属约为65.3MYA, 苎麻属约为67.5MYA, 亚麻属约为90.5MYA。主要麻类作物进化时间越久, 同属不同种之间ITS变异区段越多。
, ZHANG Li-Wu
Sequences comparison of ribosomal internal transcribed spacer (ITS) could provide evidence for the systematic classification and evolutionary relationships of main bast fiber crops and other species. In this study, the ITS sequences of 32 main bast fiber crops and 11 other species with reference genome sequences were obtained from cloning or GenBank database. The whole gene length, G+C content, and the difference of homologous percentage were analysed using MEGE software. The ITS average lengths of sequences from jute ( Corchorus), kenaf ( Hibiscus), ramie ( Boehmeria nivea) and flax ( Linum usitatissimum) were 963, 939, 658, and 686 bp, respectively. And the corresponding G+C contents were 57.87%, 58.03%, 59.05%, and 53.75%, respectively. The variation of jute ( Corchorus) concentrated on a region of 220 to 386 bp, kenaf ( Hibiscus) on two regions of 206 to 347 bp and 599 to 713 bp, ramie ( Boehmeria nivea) on four regions of 158 to 163 bp, 193 to 199 bp, 288 to 333 bp, and 681to 688 bp, and flax ( Linum usitatissimum) on five regions of 219 to 229 bp, 235 to 240 bp, 427 to 432 bp, 468 to 484 bp, and 588 to 594 bp. Phylogenetic analysis showed that jute and kenaf shared a relatively close genetic relationship while the others had a far genetic relationship, which is consistent with the relationship of traditional species classification in systematic botany. In study of comparative genomics, the genome sequecne of cotton might be regarded as a reference for kenaf or jute, and the genome sequence of Populus trichocarpa or Ricinus communis might be regarded as a reference for ramie. We deduced that the evolutionary time of kenaf, jute, ramie and flax could be roughly estimated as 33.7, 65.3, 67.5, and 90.5 million years ago, respectively, showing the longer evolution time the more variation regions of ITS in different species of bast fiber crops in the same genus.
黄麻(Corchorusspp.)、红麻(Hibiscus cannabinus)、苎麻(Boehmeriaspp.)与亚麻(Linum usitatissimum)是大面积栽培的麻类作物(以下简称主要麻类作物), 与粮、棉、油、菜并列为第五大作物群, 其种植历史可追溯到远古新石器至尧舜时代[1, 2]。根据考古证据, 大麻纺织约有6000年历史, 黄麻纺织有5000年以上历史。
我国麻类种质资源极为丰富, 其中, 苎麻、圆果种黄麻、大麻起源于中国[1, 2, 3, 4, 5]。陶爱芬等[3]通过SRAP (Sequence-related amplified polymorphism)和ISSR (Inter-simple sequence repeat)分子标记结合形态学方法对黄麻起源和演化研究认为, 非洲是世界黄麻属野生种和长果种黄麻的起源中心, 而中国南部地区是圆果种黄麻野生种和栽培种的起源中心。Zhang等[4]采用ISSR分子标记, 研究来自28个国家和地区的21份红麻野生种、半野生种和63份红麻栽培品种的遗传多样性与亲缘关系, 提出赞比亚可能为红麻野生种的起源与演化中心。张波等[5]通过对广西、云南等10省(区)的51个山区县(市)的苎麻资源考察, 发现中国的苎麻属植物多数分布于云南、广西、贵州等南方省(区), 由西南、华南向北逐渐减少, 推测广西和云南, 即云贵高原可能是中国苎麻属植物的多样性中心, 是苎麻起源地之一。亚麻原产于近东和地中海沿岸, 公元前二世纪引入栽培。目前普遍认为, 近东和地中海沿岸等地是栽培亚麻的起源地[2]。
然而, 在我国有许多植物名中带麻字。它们以草本为主, 其中供人们利用的韧皮部纤维作物主要有黄麻、红麻、苎麻、亚麻、工业大麻等, 涉及不同科、属。前人的研究主要集中在单一麻类作物起源、分布与评价等方面, 其系统位置、进化关系及与测序作物的亲缘关系尚不明确。
史全良等[6]和陈仁芳等[7]报道了桑的核糖体内转录间隔区(internal transcribed spacers, ITS)序列的多样性, 并探讨桑资源的系统位置与进化关系。这表明, ITS序列受到的选择压力较小, 在被子植物中其序列具有保守性, 适合用于低分类群的系统进化分析。本研究拟分析主要麻类作物与部分测序作物的ITS序列, 探讨其系统位置与进化关系, 旨在为其遗传进化和比较基因组学的研究提供依据。
收集43份不同植物资源的ITS序列, 分别为32份麻类植物、11份测序植物(表1)。其中, 圆果种黄麻179、长果种黄麻宽叶长果与红麻福红952来自于福建农林大学麻类遗传育种与综合利用研究室, 黄麻179、宽叶长果和福红952皆为全国麻类品种认定区域试验的对照品种。其他材料的ITS序列来自公共数据库NCBI (http://www.ncbi.nlm.nih.gov/)。
在32份麻类植物中(图1), 包括黄麻属的圆果种黄麻(Corchorus capsularis)、长果种黄麻(Corchorus olitorius)、假长果种(Corchorus pseudo-olitorius)、三室种(Corchorus trilocularis)、三齿种(Corchorus tridens)、梭状种(Corchorus fascicularis)、Corchorus siliquosus、假黄麻(Corchorus aestuans) 8种植物; 木槿属的红麻(Hibiscus cannabinus)、黄槐(Hibiscus surattensis)、木槿(Hibiscus radiates)、红叶槿(Hibiscus acetosella)、H. sabdariffavar. sabdariffa、变种玫瑰茄(Hibiscus sabdariffavar.altissima) 6种植物; 苎麻属的苎麻(Boehmeria nivea)、青叶苎麻(Boehmeria niveavar.tenacissima)、大叶苎麻(Boehmeria longispica)、细野麻(Boehmeria gracilis)、悬铃叶苎麻(Boehmeria tricuspis)、赤麻(Boehmeria silvestrii)、长叶苎麻(Boehmeria penduliflora)、束序苎麻(Boehmeria siamensis)、帚序苎麻(Boehmeria zollingerianavar. blinii)、密球苎麻(Boehmeria densiglomerata)、滇黔苎麻(Boehmeria pseudotricuspis)、阴地苎麻(Boehmeria umbrosa) 12种植物; 亚麻属的亚麻(Linum usitatissimum)(该植物同时属于测序植物)、野亚麻(Linum stelleroides)、宿根亚麻(Linum perenne)、短柱亚麻(Linum pallescens) 4种植物; 还包括其他麻类植物大麻(Cannabis sativa)(该植物同时属于测序植物)与蕉麻(Musa textilisNee)。
 | 图1 世界上主要的麻类作物田间图(部分图片来自网站)Fig. 1 Field pictures of main bast fiber crops in the world (Some pictures from website) |
在11份测序植物中, 有参考基因组序列的植物有拟南芥(Arabidopsis thaliana)[8]、陆地棉(Gossypium hirsutum)[9]、雷蒙德氏棉(Gossypium raimondii)[10]、杨树(Populus trichocarpa)[11]、可可(Theobroma cacao)[12]、大豆(Glycine max)[13]、蒺藜苜蓿(Medicago truncatula)[14]、麻疯树(Jatropha curcas)[15]、蓖麻(Ricinus communis)[16]、葡萄(Vitis vinifera)[17]和水稻(Oryza sativa)[18]。
采用改良的CTAB法[19]提取红麻与黄麻的DNA。用Nanodrop紫外分光光度计检测DNA的纯度与浓度, 将DNA的浓度稀释至50 ng μ L-1, 保存于-20℃, 用于PCR扩增。
ITS序列扩增左右引物分别为18S For: 5° -TGG GGATAGATCATTGCAATTGTTGGTC-3° 和28S Rev: 5° -TCCYGGTTCGCTCGCCGTTACTA-3° (Y = A/T), 由生工生物工程(上海)股份有限公司合成。PCR 体系15 µ L, 包括DNA模板3 µ L、10 mmol L-1上下游引物各1.0 µ L、10× PCR缓冲液1.5 µ L、25 mmol L-1 MgCl2 1.2 µ L、10 mmol L-1 dNTPs Mix 0.3 µ L、5 U µ L-1Taq酶(MBI Ferments) 0.2 µ L、ddH2O 6.8 µ L, 再加10 µ L矿物油覆盖。扩增程序为95℃ 3 min; 95℃ 30 s, 52℃ 30 s, 72℃ 45 s, 共35个循环; 72℃延伸7 min, 4℃保温。扩增产物经1%琼脂糖凝胶电泳检测, 用DNA纯化回收试剂盒(TIANGEN)回收后送铂尚生物技术有限公司测序, 获得DNA序列。
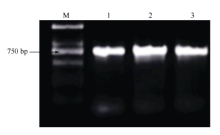
对圆果种黄麻黄麻179、长果种黄麻宽叶长果与红麻福红952进行扩增(图2), 获得ITS序列分别为957、958与941 bp (表1)。根据GenBank公布的ITS序列信息, 下载获得其他40份植物的ITS序列, 包含亚麻(Linum usitatissimum)、苎麻(Boehmeria nivea)、大麻(Cannabis sativa)与蕉麻(Musa textilisNee)等29份麻类作物(表1)。
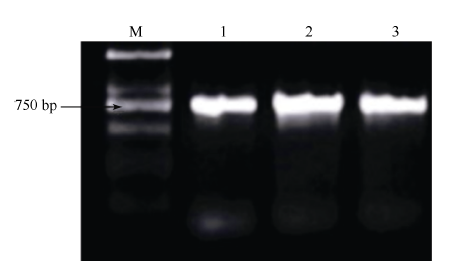 | 图2 ITS序列扩增电泳图Fig. 2 Electrophoregram of ITS 1: 圆果种黄麻黄麻179; 2: 长果种黄麻宽叶长果; 3: 红麻福红952; M: DL2000。 1: Corchorus capsularis Huangma 179; 2: Corchorus olitorius Kuanyechangguo; 3: Hibiscus cannabinus Fuhong 952; M: DL2000 marker. |
| 表1 主要麻类作物与相关测序植物的ITS长度及GC含量变异 Table 1 ITS sequence length and GC contents (bp, %) in main bast fiber crops and related species with reference genome sequences |
通过比对, 确定本研究涉及材料ITS序列的18S rRNA基因3′ 端、5.8S rDNA基因、28S rRNA基因5′ 端的碱基序列(表1), 发现不同植物之间ITS序列有不同程度变异。
在主要麻类作物中, 黄麻属ITS基本序列全长963 bp, G+C含量57.87% (各序列长度为8份黄麻属植物ITS序列的平均值)。其中, ITS1序列长度为277 bp, G+C含量为60.49%; ITS2序列长度为232 bp, G+C含量60.89%; 5.8S序列长度为162 bp, G+C含量54.86%。黄麻属ITS序列的同源性变化在93.12%至97.64%之间。序列比对结果表明, 变异区域集中在1个区段, 即220~386 bp之间。
红麻属ITS基本序列全长939 bp, G+C含量58.03% (各序列长度为6份红麻属植物ITS序列的平均值)。其中, ITS1序列长度为254 bp, G+C含量59.39%; ITS2序列长度为223 bp, G+C含量63.20%; 5.8S序列长度为 163 bp, G+C含量54.10%。ITS序列的同源性变化在91.23%~99.16%之间。序列比对结果表明, 变异区域集中在2个区段, 即206~347 bp和599~713 bp。
苎麻属ITS基本序列全长658 bp, G+C含量59.05% (各序列长度为12份苎麻属植物ITS序列的平均值)。其中, ITS1序列长度为177 bp, G+C含量58.90%; ITS2序列长度为237 bp, G+C含量65.11%; 5.8S序列长度为160.63%, G+C含量52.02%。ITS序列的同源性变化在92.19%至100.00%之间。序列比对结果表明, 变异区域分布在4个区段, 即158~ 163 bp、193~199 bp、288~333 bp和681~688 bp。
亚麻属ITS基本序列全长686 bp, G+C含量53.75%(各序列长度为4份亚麻属植物ITS序列的平均值)。其中, ITS1序列长度为213 bp, G+C含量56.81%; ITS2序列长度为205 bp, G+C含量53.99%; 5.8S序列长度为169 bp, G+C含量51.48%。ITS序列的同源性变化在86.63%~97.75%之间。序列比对结果表明, 变异区域分布广泛, 分别在5个区段, 即219~229 bp、235~240 bp、427~432 bp、468~484 bp和588~594 bp。
比较43份植物ITS序列, 得出其同源性(percent identity)的平均值为96%, 其中葡萄与水稻同源性最低, 仅为83.54% (表2)。表明供试材料的ITS序列都具有一定的保守性。应用软件MEGE 5.2确定本研究各材料的系统位置(图2)。将水稻(Oryza sativa)作为外类群被单独分出, 其余植物则被分为8个分支。
| 表2 43份植物ITS序列同源性比较 Table 2 Comparison of similar identity of ITS sequences in 43 plants |
依次分析发现, 第1支仅包括葡萄(Vitis vinifera), 第2支仅包括蕉麻(Musa textilisNee), 表明蕉麻在以麻字命名的植物中可单独分出。
第3支包括可可(Theobroma cacao)、蒺藜苜蓿(Medicago truncatula)、大豆(Glycine max)、大麻(Cannabis sativa)和亚麻属4个种。表明在以麻字命名的植物中, 大麻与亚麻属植物的亲缘关系较近。
第4支包括苎麻属的12个不同种, 其靴带支持率为100% (即可靠性为100%)。进一步细分, 这12个不同种可划分4个小分支。其中, 苎麻(Boehmeria nivea)与青叶苎麻(Boehmeria nivea var. tenacissima)之间的靴带支持率为100%, 这二者的形态虽有不同, 但用途却基本相似, 在苎麻属分类中均为苎麻组。大叶苎麻(Boehmeria longispica)、细野麻(Boehmeria gracilis)、悬铃叶苎麻(Boehmeria tricuspis)与赤麻(Boehmeria silvestrii)间的靴带支持率也达到了100%, 均为大叶苎麻组, 皆具有无融合生殖能力。这种能力可以保持物种的遗传特性, 通过基因导入或杂交等方式, 转育到栽培种中, 在杂交育种与杂种优势上有很大的发展空间。
第5支包括麻疯树(Jatropha curcas)、蓖麻(Ricinus communis)和杨树(Populus trichocarpa), 靴带支持率为95%。说明在以麻字命名的植物中, 蓖麻、麻疯树与杨树的亲缘关系较近。联合第4、第5支来看, 在测序植物中, 蓖麻或杨树与苎麻属的亲缘关系较近。
第6支仅包括拟南芥(Arabidopsis thaliana)。第7支包括陆地棉(Gossypium hirsutum)、雷蒙德氏棉(Gossypium raimondii)和红麻属6个不同种。表明对红麻进行比较基因组学研究时, 可选择雷蒙德氏棉或陆地棉作为参考基因组。
第8支包括黄麻属8个不同种, 其靴带支持率为100%。其中, 假黄麻C. aestuans可能是黄麻属栽培种圆果种黄麻、长果种黄麻、假长果种(Corchorus pseudo-olitorius)以及三室种(Corchorus trilocularis)的共同祖先。联合第7、第8支来看, 在测序植物中, 棉花与黄麻属的亲缘关系较近。表明黄麻也可以选择雷蒙德氏棉或陆地棉作为参考基因组。
比较主要麻类作物在整个进化系统中位置, 推测黄麻与红麻的亲缘关系较近, 为一大分支。亚麻属与大麻亲缘关系较近, 为一大分支。而苎麻属单独分出。
从表3可以看出, 不同植物的进化距离平均值为0.171, 变异系数为42.45%。其中, 蒺藜苜蓿与水稻之间的进化距离最高, 为0.480。以雷蒙德氏棉为参考, 发现雷蒙德氏棉与红麻的进化距离为0.020, 与长果种黄麻为0.193, 与圆果种黄麻为0.198, 与蓖麻为0.256, 与大麻为0.233, 与亚麻为0.273, 与苎麻为0.241, 与蕉麻为0.279。这进一步验证黄麻、红麻与亚麻、苎麻的亲缘关系较远。
根据测序植物的进化时间[10], 推测主要麻类植物的进化时间。红麻属约为33.7百万年前(million years ago, MYA), 黄麻属约为65.3 MYA, 苎麻约为67.5 MYA, 亚麻约为90.5 MYA。结合主要麻类作物ITS变异区域和进化时间可以看出, 植物进化时间越久, 同属不同种的ITS序列变异区段越多。
对于用ITS序列分析不同植物之间的系统位置与进化关系, 已有一些报道[6, 7], 但对于主要麻类作物的系统位置剖析却未见报道。目前, 大面积栽培的韧皮部纤维麻类作物有红麻、苎麻、黄麻、亚麻与工业大麻, 各属不同科、属。红麻(Hibiscus cannabinus L.)是锦葵科(Malvaceae)木槿属(Hibiscus L.)一年生韧皮部纤维作物, 亦称洋麻、槿麻; 属短日照作物, 能适应冷热干湿等各种气候条件[22, 23]。苎麻[Boehmena nivea (L.) Gaud.]为荨麻科(Uricaeeae)苎麻属(Boehmena)多年生宿根性草本植物, 又名银苎、苎根、山麻、天青地、白草等, 是一种重要的韧皮纤维作物[24, 25]。黄麻为椴树科(Tiliaceae)黄麻属(Corchorus)一年生的韧皮纤维作物, 黄麻属有约40个种, 具有栽培价值的有圆果种(C. capsularis)和长果种(C. olitorius)[26, 27]。亚麻(Linum usitatissimum L.)为金虎尾目亚麻科(Linaceae)亚麻属(Linum)一年生草本植物, 是古老的纺织纤维作物和油料作物, 按其经济性状和用途可分为油用、纤维用和油纤兼用三种类型[28]。
按恩格勒被子植物分类系统观点, 主要麻类作物属于被子植物, 是单元起源的。苎麻为荨麻目, 属于同被花类原始花被亚纲; 亚麻(Linum usitatissimum L.)属金虎尾目; 红麻与黄麻为锦葵目, 属于异被花类。哈钦松被子植物分类系统观点认为, 毛莨目为被子植物的原始类群, 单子叶植物起源于毛莨目。按双子叶植物进化时间来划分, 锦葵目晚于金虎尾目与荨麻目。这与本研究推测主要麻类作物的进化时间是一致的, 即红麻和黄麻的进化时间要晚于亚麻与苎麻。
本研究对43份植物ITS序列分析发现, 虽各材料ITS序列有不同程度的变异, 但主要麻类作物ITS序列具有一定的保守性。在主要麻类作物中, 黄麻与棉花亲缘关系较近, 红麻与棉花亲缘关系更近。而雷蒙德氏棉具有参考基因组序列, 基因组大小为775.2 Mb[10], 这将为红麻、黄麻的遗传进化和比较基因组学研究提供便利。Zhang等[29]对红麻旺盛生长期的根、茎、叶、茎皮和茎骨进行转录组测序, 发现91.01%雷蒙德氏棉基因与红麻存在共线性。在黄麻比较基因组学研究中, Biswas等[30]以陆地棉为参考基因组, 进行黄麻染色体定位, 发现黄麻7条染色体分别与陆地棉的第1至第8染色体存在一定共线区段。Kundu等[31]以雷蒙德氏棉为参考基因组, 发现与黄麻有29.2%的共线区段。基于系统进化树, 我们认为假黄麻应是长果种黄麻与圆果种黄麻的共同祖先, 这与Kundu等[32]基于形态学和微卫星标记的黄麻分类结果较一致。而苎麻属划分为另一分支, 从分支图来看, 与测序植物杨树、蓖麻和麻疯树的亲缘关系较近。Chen等[33]对苎麻茎皮进行转录组测序, 发现苎麻与葡萄、蓖麻和大豆的亲缘关系较近。而Liu等[34]也对苎麻茎皮进行转录组测序, 发现苎麻与葡萄、蓖麻与杨树亲缘关系较近。结合本研究的结果, 在比较基因组学研究中, 苎麻可参考蓖麻或杨树。蓖麻和杨树均已测序, 其参考基因组大小分别为350 Mb[16]和480 Mb[35]。在苎麻属不同种的系统进化研究中, 我们认为苎麻组与大叶苎麻组亲缘关系更加紧密。这与前人研究发现苎麻组与大叶苎麻组亲缘关系近[36, 37, 38, 39]的研究结果是一致的。最近, Yu等[39]通过ITS等序列把31份苎麻属植物划分为四组, 该文IV、II、I三组与本研究中苎麻组、大叶苎麻组与其余苎麻组相吻合。在主要麻类作物中, 亚麻属植物与大麻为一小支, 两者皆有参考基因组序列[40, 41], 大麻[40]与亚麻[41]参考基因组大小分别为534 Mb和373 Mb, 这些将有助于韧皮部纤维作物的遗传基础研究。
主要麻类作物的ITS序列具有一定保守性, 可用于不同麻类植物的系统进化分析。主要韧皮部纤维植物中, 黄麻属、红麻属的亲缘关系近, 而黄麻属、红麻属与亚麻属、苎麻属的亲缘关系较远。在比较基因组学研究, 红麻、黄麻可参考棉花, 苎麻可选择杨树或蓖麻。推测红麻属、黄麻属、苎麻属、亚麻属的进化时间分别为33.7、65.3、67.5、90.5百万年前(million years ago, MYA)。
The authors have declared that no competing interests exist.
作者已声明无竞争性利益关系。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|