

{kind=link}
{kind=link}
利用90k芯片技术进行小麦穗部性状QTL定位
[武炳瑾, 简俊涛, 张德强, 马文洁, 冯洁, 崔紫霞, 张传量, 孙道杰*  ]
]
]
|
|


第一作者联系方式: E-mail: wbj2010@163.com
小麦穗部性状与产量密切相关, 挖掘穗部性状基因及其关联分子标记具有重要意义。本研究以周8425B×小偃81衍生的RIL群体(F8)为材料, 利用90k芯片标记构建的高密度遗传图谱对3个环境下的穗长、小穗数、不育小穗数、穗粒数、千粒重进行QTL定位。共检测到19条染色体上的71个QTL, 变异解释率(PVE)范围为2.10%~45.25%, 其中37个位点为主效QTL (PVE>10%)。 QSl.nafu-6A.2 (穗长)、 QSl.nafu-7A (穗长)、 QSsn.nafu-2A.1 (不育小穗数)、 QSsn.nafu-2D (不育小穗数)和 QGns.nafu-2B (穗粒数)在多个环境中被检测到, 且LOD>10, PVE>20%。位于同一个基因簇中的 QSl.nafu-6A.2(穗长)、 QGns.nafu-6A (穗粒数)和 QTgw.nafu-6A (千粒重)在多个环境中被检测到, 且与已报道的相关位点位置相同或相近, 在分子标记辅助育种中具有较大参考价值。
Spike traits are important to grain yield in wheat. Molecular markers associated with genes/QTLs controlling spike traits are highly valuable to marker-assisted breeding. A recombinant inbred line (F8) population derived from Zhou 8425B ×Xiaoyan 81 were evaluated in three environments, and QTLs for spike length, spikelet number per spike, sterile spikelet number, grain number per spike and thousand-grain weigh were mapped into a high-density genetic map built by 90k chip. A total of 71 QTLs were located on 19 chromosomes, and the phenotype variation explained (PVE) by a single locus ranged from 2.10% to 45.25%. Thirty-seven loci were considered as main-effect QTLs owing to the PVE larger than 10%. QTLs QSl.nafu-6A.2 for spike length, QSl.nafu-7A for spike length, QSsn.nafu-2A.1 for sterile spikelet number, QSsn.nafu-2D for sterile spikelet number and QGns.nafu-2B for grain number per spike were identified repeatedly in different environments with the LOD value higher than 10 and PVE larger than 20%. QSl.nafu-6A.2 for spike length, QGns.nafu-6A for grain number per spike and QTgw.nafu-6A for thousand-grain weight were mapped in a cluster on chromosome 6A and might be applicable in marker-assisted selection because they have been detected in multiple environments and close to the loci reported.
小麦穗部性状是产量的重要构成要素, 对控制穗粒数、千粒重等主要穗部性状进行QTL定位, 明确其在染色体上的位置和效应, 对产量性状的遗传改良具有重要意义。迄今, 已报道了200多个控制小麦穗部相关性状的QTL, 分布在小麦21条染色体上[1, 2, 3, 4, 5]。卢翔等[6]利用F2群体将穗长、小穗数、穗粒数相关QTL定位到1A、2A、5B、5D染色体上; Wang等[7]利用F2:3群体在4BL、5A、6A染色体上定位了控制穗粒数、可育小穗数、穗长、小穗数的QTL。Li等[8]检测到46个影响籽粒产量、千粒重、穗粒数、有效穗数、可育小穗数、不育小穗数和总小穗数的QTL。
遗传连锁图谱的密度直接关系到QTL定位的准确性和可信度。SNP标记在小麦基因组中数量大且分布广泛, 基于Illumina技术平台的小麦90K全基因组SNP芯片已被广泛应用于小麦遗传连锁图谱的构建、DNA指纹分析、群体结构和连锁不平衡分析以及基因定位等领域[9]。本研究利用90k芯片标记构建的高密度遗传连锁图谱, 对穗部性状进行QTL定位, 为小麦的分子标记辅助育种、分子聚合育种以及基因克隆提供依据。
使用周8425B和小偃81构建的含有102个家系的F8重组自交系(RIL)群体作为试验对象。周8425B是河南省周口市农业科学院创制的核心亲本, 具有配合力较好、抗病性强、矮秆、大穗、大粒等优点; 小偃81是李振声院士选育的高产、优质、多穗型品种。
试验材料于不同年份分别种植于陕西杨凌(2013— 2014, E1)、河南安阳(2014— 2015, E2)和陕西杨凌(2015— 2016, E3), 随机区组设计, 小区行长2 m, 行距0.23 m, 2次重复。常规田间管理, 生长期间未发生严重病虫害和倒伏现象。
小麦成熟后, 参照《小麦种植资源描述规范和数据标准》[10], 从每个小区随机选10个单株调查穗长、穗粒数、小穗数、不育小穗数和千粒重, 取平均值用于表型及遗传分析。
使用Microsoft Excel统计分析数据, 使用IciMaping软件中的ANOVA功能计算遗传力。
使用Illumina公司的小麦90k芯片进行全基因组扫描, Genomestudio v1.0软件分型, 筛选亲本差异标记并用于高密度遗传图谱的构建。
使用基于最大似然估计法的CarthaGè ne构建遗传连锁图谱, 取LOD=9.7, 阈值设为30 cM。分组后使用greedy和annealing功能调整并寻找最佳图谱。使用MapChart软件绘制遗传连锁图谱。
使用基于逐步回归的完备复合区间作图法IciMapping[11]中的BIP功能进行多环境QTL定位, 步移速度为1.0 cM, P临界值为0.001, 使用LOD=2.5作为检测阈值。
在3个检测环境中, 母本周8425B在穗长、小穗数、千粒重上大于父本小偃81, 而父本在穗粒数上大于母本, 但在E3环境母本的穗粒数略大于父本, 与其他2个环境不一致。不育小穗数两亲本相差不大, 各年际间环境不同, 略有增减。5个穗部性状的偏度和峰度绝对值均小于1, 符合正态分布。各性状在所有环境下都存在双向超亲分离现象, 表明为多基因控制的数量性状, 适于进行QTL定位。穗长和小穗数的变异系数都大于10, 可能是亲本间差异较大导致后代发生了更广泛的分离, 说明有很大的改良潜力。5个穗部性状中, 穗粒数的遗传力最小, 为0.57, 穗长的遗传力最大, 为0.82, 表明穗长主要受基因控制, 受环境影响较小(表1)。
| 表1 穗部性状表型分析 Table 1 Phenotypic analysis of panicle traits |
相关分析显示, 穗长与小穗数、不育小穗数呈正相关, 与千粒重呈负相关; 小穗数与不育小穗数呈正相关, 与千粒重呈负相关; 穗粒数与不育小穗数呈负相关(表2)。
| 表2 小麦穗部性状相关系数 Table 2 Correlation coefficientsof panicle traits |
本实验使用的90k芯片共有81 588个检测探针, 覆盖小麦全基因组。其中检测到亲本多态性位点11 037个, 控制缺失率(0.05)并去除不连锁标记后共9290个标记用于遗传图谱构建。所有SNP标记共构建了49个连锁群, 覆盖小麦21条染色体, 遗传图谱总长3894.64 cM, 平均标记密度0.42 cM。
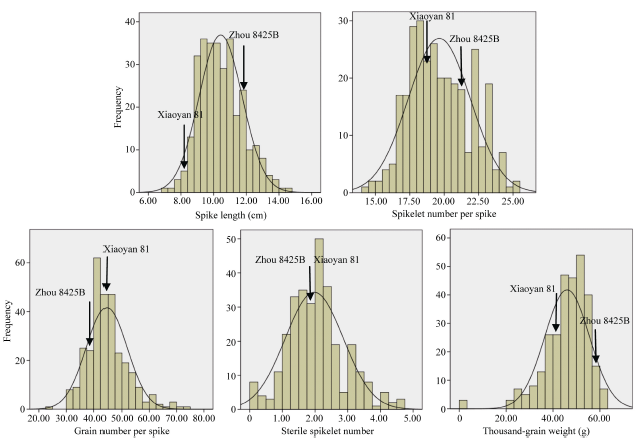 | 图1 穗部性状表型分析(图中包含3个环境全部数据。)Fig. 1 Phenotypic analysis of panicle trait(The figure contains all the data in 3 environments.) |
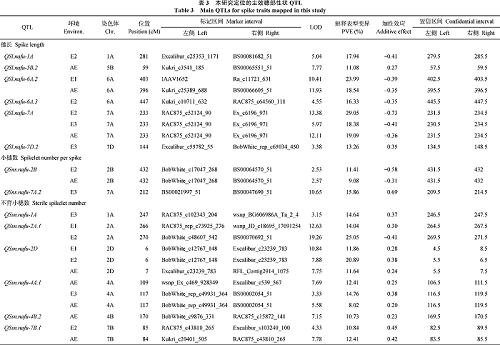
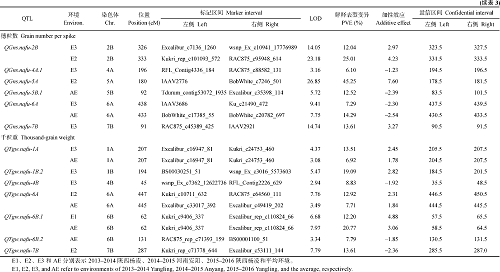
利用3个环境及其平均值的表型数据, 共定位71个穗部性状相关的QTL, 分布在19条染色体上, 控制穗长(18个)、千粒重(16个)、不育小穗数(14个)、小穗数(12个)和穗粒数(11个), 其中37个位点对表型变异的解释率(PVE)超过10%, 为主效QTL。有11个位点在2个或者2个以上环境中被检测到(表3和图2)。
 | 图2 4A、6A和7B染色体上定位的穗部性状QTL(穗长、小穗数、不育小穗数、穗粒数和千粒重相关QTL分别用黑、红、绿、蓝和棕色标注。)Fig. 2 QTLs for spike traits on chromosomes 4A, 6A, and 7B(QTLs for spike length, spikelet number per spike, sterile spikelet number, grain number per spike, and thousand-grain weight were marked in black, red, green, blue, and brown, respectively.) |
18个控制穗长和QTL分布在1A、1D、2B、2D、3A、3B、4A、5B、6A、6B、7A、7B和7D染色体上, 其中9个为主效QTL (PVE> 10%)。QSl.nafu-6A.2和QSl.nafu-7A在多个环境中被检测到, 其LOD值分别为10.41和13.38, PVE值分别为23.99和29.05, 增加穗长的效应其均来自小偃81。
12个控制小穗数的QTL位于1A、2A、2B、4B、4D、6A、6D、7A、7B、7D染色体上, 其中7个是主效QTL。QSns.nafu-2B在2个环境的同一位置被检测到, 是较稳定QTL, 其加性效应来自小偃81。
15个控制不育小穗数的QTL分布于1A、2A、2D、4A、4B、4D、5A、7A和7B染色体上, 包括10个主效QTL, 单个位点的PVE在10.30%~25.05%范围内。QSsn.nafu-4A.1、QSsn.nafu-7B.1和QSsn.nafu-2D在多环境中被检测到。
11个控制穗粒数的QTL位于2B、4A、5A、5B、6A、6D、7A、7B和7D染色体上, 其中6个是主效QTL。QGns.nafu-2B和QGns.nafu-6A在2个环境中被检测到, 其他QTL只在一个环境中检测到。
16个控制千粒重的QTL分布于1A、1B、2B、2D、3A、3B、4B、6A、6B、7B染色体上, 其中8个是主效QTL, 其PVE范围为12.20%~20.77%。QTgw.nafu-1A、QTgw.nafu-6A和QTgw.nafu-6B.1在2个环境中被检测到, 其加性效应来自周8425B。
普通小麦(Triticum aestivum L.)是一个具有A、B、D染色体组的异源六倍体物种, 长期进化和人工选育使小麦资源的遗传多样性水平偏低且遗传基础狭窄, D染色体组表现尤为突出, 大部分学者认为3个基因组等位变异丰富度为B> A> D[12, 13, 14], 而Roussel等[15]则报道A> D> B。本研究结果与大部分学者一致, 认为B基因组的遗传多样性最高, A基因组次之, D基因组最低。A、B、D基因组所占比例分别是34.80%、55.76%和9.44%, 以1B染色体标记覆盖数最高, 4D染色体覆盖最少。
| 表 3 Table 3 |
| 表 3(续表) Table 3 |
不同染色体组标记数分布不平衡的可能原因, 一是与基因组来源有关, 六倍体小麦的B基因组起源于异花授粉的山羊草属, 而A基因组的供体乌拉尔图小麦和D基因组供体粗山羊草均为自花授粉植物, 通常异花授粉植物的遗传多样性都高于自花授粉植物[16]; 在小麦进化过程中A、B基因组形成了较多的四倍体种, 如考尔希小麦、波斯小麦、圆锥小麦、波兰小麦等, 这些亲缘种均可能形成六倍体小麦, 从而丰富了A、B基因组的遗传多样性, 而D基因组与A、B基因组间均无四倍体形成, 减少了D基因组间的基因交流, 从而限制了遗传多样性的产生。二是与自然和人工选择有关, D基因组可能携带控制普通小麦适应性、抗逆性和加工品质等重要性状的基因[17], 在长期的选择和育种过程中所承受的选择压力大于A和B基因组, 从而造成更多、更强的选择牵连效应发生, 导致其遗传多样性较低。小麦D基因组的草图已完成, 研究发现, 正是由于D基因组的加入, 才使小麦的抗病性、适应性与品质得到大大改良[17]。
业已证明, 在不同群体和环境下所定位的同一性状的某些QTL具有很好的一致性, 尤其是一些效应大的QTL。定位区间经过相互验证的QTL为进一步鉴定和克隆相关基因奠定了基础。本研究共定位到71个QTL, 其中11个在2个或者2个以上环境中被检测到, 是较稳定的QTL, 其他QTL只能在一个环境中检测到。其原因可能是穗部性状受多基因控制且易受环境影响, 穗部性状基因之间也存在相互作用, 所以部分QTL在多环境检测中表现不稳定或环境特异表达。
控制穗长的QSl.nafu-5B.2包含在已报道的QSl.cau- 5B[18]标记区间内, 本研究将其标记区间缩小至2 cM范围内; QSl.nafu-7D.2与高尚等[19]定位的QSL.SAU-7D.1有共同标记BobWhite_rep_c65034_450, 7D染色体上控制穗长的基因很有可能位于该标记附近; QSl.nafu-7A与已报道的QSl.hwwgr-7AL.2[20]位置相近, 并且能够在3个环境中被检测出来, 其PVE高达29.05%, 是一个稳定的主效QTL。
控制小穗数的QSns.nafu-2B在2个环境下被定位在同一位置, 是一个较稳定的QTL; 4B染色体上的QSsn.nafu- 4B.2与已报道的QSSN4B.4-17[21]位置相近。3个控制不育小穗数的主效QTL被重复检测到, 其中QSsn.nafu-4A.1和QSsn.nafu-7B.1在2个环境中被检测到, QSsn.nafu-2D在3个环境中被定位在同一位置。前人也在7B染色体上定位到控制不育小穗数的QTL[22], 因使用的标记类型不同, 所以无法判断与本研究结果的一致性。此外, 本研究首次在4A和2D染色体上定位到控制不育小穗数的主效QTL QSsn.nafu-4A.1和QSsn.nafu-2D。
控制穗粒数的QGns.nafu-4A.1与已报道的QGns.cau- 4A[18]位置接近; QGns.nafu-5B.1与QGns.cau-5B.2[18]的区间部分重叠; QGns.nafu-6A包含在QKnps.hwwgr-6AL[23]的标记区间内。
控制千粒重的QTgw.nafu-4B包含在已报道的QGW4B.4-17[21]的标记区间内, 并且与QTkw.hwwgr- 4BS[23]有共同标记; 本研究在2个环境中检测到QTgw.nafu-6A, 与刘凯等[21]和Li等[23]在6A染色体上鉴定的QTL有共同区间; 吴秋红等[18]在3个环境中检测到QGws.cau-6B.2, 与本研究定位到的QTgw.nafu-6B.2存在共同区段; 本研究在2个环境中检测到表型贡献率为20.77%的QTgw.nafu-6B.1, 该位点未见报道, 可能是一个新的控制千粒重的主效QTL。
相关性状的QTL往往存在于相同或相近染色体区段[24]。本研究发现4A、6A、7B染色体上存在QTL富集现象(图2), 在4A染色体上聚集了控制穗长、不育小穗数和穗粒数的QTL; 在6A染色体上定位了控制穗长、小穗数、不育小穗数、穗粒数、千粒重的QTL; 7B染色体上检测到控制穗长、小穗数、千粒重的QTL位点, 几乎位于同一位置, 也有可能为同一位点控制多个表型性状。张坤普等[25]在4A染色体上发现与穗长和穗粒数相关的QTL簇; 丁安明等[26]和刘凯等[21]也在6A染色体上检测到控制千粒重、穗粒数和小穗数的QTL簇。利用这些“ 一因多效” 或紧密连锁QTL, 结合稳定表达的位点, 如QSl.nafu-6A.2、QSl.nafu- 7A、QSsn.nafu-4A.1、QGns.nafu-6A、QTgw.nafu-6A等, 通过回交和分子标记辅助选择, 可同时聚合控制多个性状不同位点的等位基因, 从而创制出优异的小麦种质资源。
The authors have declared that no competing interests exist.
作者已声明无竞争性利益关系。The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|