


{kind=link}
{kind=link}
{kind=link}
{kind=link}
抗小麦黄矮病相关蛋白激酶TiDPK1与BYDV外壳蛋白的互作
引用本文
汪信东, 陈亮, 张增艳. 抗小麦黄矮病相关蛋白激酶TiDPK1与BYDV外壳蛋白的互作. 作物学报, 2013, 39(10): 1720-1726
[WANG Xin-Dong, CHEN Liang, ZHANG Zeng-Yan. Interaction between Wheat Resistance-related Kinase TiDPK1 and BYDV Coat Protein. Acta Agronomica Sinica, 2013, 39(10): 1720-1726]
Permissions
[WANG Xin-Dong, CHEN Liang, ZHANG Zeng-Yan. Interaction between Wheat Resistance-related Kinase TiDPK1 and BYDV Coat Protein. Acta Agronomica Sinica, 2013, 39(10): 1720-1726]
Copyright©2013, Editorial office of Acta Agronomica Sinica
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
抗小麦黄矮病相关蛋白激酶TiDPK1与BYDV外壳蛋白的互作
摘要
小麦黄矮病是由大麦黄矮病毒(
关键词:
小麦; 蛋白激酶TiDPK1; 大麦黄矮病毒外壳蛋白; 酵母双杂交; 双分子荧光互补; 蛋白互作
Interaction between Wheat Resistance-related Kinase TiDPK1 and BYDV Coat Protein
Abstract
Yellow dwarf virus disease is one of the important diseases of wheat (
Keyword:
Wheat; Protein kinase TiDPK1; Coat protein of Barley yellow dwarf virus; Yeast two-hybrid: Bimolecular fluorescence complementation; Protein-protein interaction
引言
黄矮病是由大麦黄矮病毒( Barley yellow dwarf virus, BYDV)引起的小麦病毒病, 在世界各地小麦产区均有发生, 我国西北、华北、西南、华东等近20个省、市、自治区的冬春麦区每年都有不同程度的病害, 造成小麦减产5%~30%, 个别严重地块减产幅度超过50%[1], 目前除抗病品种外, 尚无其他有效的防治措施。优良的抗病资源是抗病育种的关键之一。小麦栽培品种中缺乏对黄矮病的有效抗源, 仅在少数材料中发现1个耐病基因[2]。偃麦草属( Thinopyrujm)、冰草属( Agropyron)、赖草属( Leymus)、披碱草属( Elymus)、鹅冠草属( Roegneria)中十余种小麦近缘植物对黄矮病免疫, 其中以中间偃麦草( Th. intermedia)被研究利用最多。国内外几个研究组分别将携带抗病基因的中间偃麦草染色体或染色体片段导入小麦背景,育成抗黄矮病的小麦异源染色体 系[3,4,5]。小麦-中间偃麦草易位系YW642, 携带源自中间偃麦草7X(7Ai-1)染色体长臂的抗黄矮病基因 Bdv2[6], 抗BYDV的PAV-Aus、-MAV、-GAV、-GPV等多个株系。
BYDV是一类正单链RNA病毒, 通过蚜虫传播, 可侵染小麦、大麦、燕麦、禾草等上百种植物。BYDV与介体蚜虫间存在一定的专化性, 每种株系只能由1种或少数几种蚜虫传播。目前, 已将BYDV-PAV、-PAS、和-GAV株系划分为 Luteovirus属, BYDV- RPV、-GPV株系划分为 Polerovirus属, 而BYDV-SGV和-RMV 株系在 Luteovirus病毒科中尚未指定种属[7]。其中PAV、PAS是欧美国家小麦黄矮病的主流株系[8], GPV主要分布于我国及瑞典[9], GAV是我国黄矮病的主流株系[10]。近年来, 国内外分别完成了BYDV-PAV[11,12]、-MAV[11]、-RPV[13]、-GAV[14]和-GPV[15]等株系的基因组测序。序列分析表明, BYDV-GAV基因组包含6个开放阅读框(open reading frame, ORF), 分别编码P1、病毒外壳蛋白(coat protein, CP)、运动蛋白(movement protein, MP)、依赖RNA的RNA聚合酶(RNA-dependent RNA polymerase, RdRp)、传毒蛋白(aphid transmission protein)和RNA沉默抑制子(P6)[16]; BYDV-PAV基因组也包含6个ORF, 分别编码P1、RdRp、传毒蛋白、CP、MP和RNA沉默抑制子(P6), 但不同地区的PAV序列差异较大。BYDV的CP可能是该病毒感染小麦的重要致病因子(与王锡锋研究员和刘艳博士的私人通讯)。
近年研究发现, 蛋白激酶识别病原效应因子或传递下游信号参与防御反应。水稻抗白叶枯病基因 Xa21编码受体类蛋白激酶(RLK)、使野生稻具有抗 Xanthomonas oryzae pv. oryzae的性质[17]; 在马铃薯抗 Pseudomonas syringae反应中, 蛋白激酶Pto能识别病原菌的效应因子(AvrPto与AvrPtoB), 并通过与抗病蛋白Prf互作共同抵御病原菌[18]; 簇毛麦抗白粉病重要基因 STK-V编码丝氨酸和苏氨酸蛋白激酶基因, 为 Pm21基因簇的关键成员, 使小麦-簇毛麦易位系具有白粉病抗性[19]。 TiDPK1是张增艳课题组从抗黄矮病易位系YW642中分离的一个蛋白激酶编码基因, 定位于易位的中间偃麦草染色体片段上。Virus-induced gene silencing (VIGS)功能分析表明, TiDPK1是小麦-中间偃麦草易位系防御BYDV反应所需基因(未发表), 但其是否与BYDV CP互作目前还不知道。
研究蛋白相互作用的方法主要有酵母双杂交(yeast two-hybrid, YTH)、双分子荧光互补(bimolecular fluorescence complementation, BiFC)、免疫共沉淀(coimmunoprecipitation, CO-IP)及pull- down等。YTH的原理主要是基于对酵母转录因子GAL4性质的认识, 即将用于互作研究的2个基因分别与GAL4 N-端的DNA结合域(DNA binding domain, BD)或GAL4 C转录激活域(activation domain, AD)融合、构建在表达载体上, 共同在特别改造的酵母菌株如AH109(GAL4-)中表达融合蛋白。若2个融合蛋白互作, 功能重建的GAL4则能激活下游报告基因的表达, 使改造酵母菌株在各种缺陷型培养基上正常生长。因此在YTH实验中, 可通过对特异改造酵母菌株在各种缺陷型培养基上生长状况及X-α-gal颜色反应等来判定蛋白间的相互关系[20]。双分子荧光互补是2002年由Hu等[21]报道的用于直观、快速判断目标蛋白在活细胞中的定位及其互作的技术。该技术巧妙地将荧光蛋白分子的发光基团分成2个互补片段, 分别与其中一个目标蛋白融合表达, 共同转化细胞, 如果荧光蛋白恢复活性、表达荧光, 则表明2个目标蛋白具有相互作用。
本研究通过YTH和BiFC技术, 研究TiDPK1与BYDV-GAV、-PAV的CP间互作关系, 有助于探究和理解 TiDPK1抗BYDV的作用机制, 对植物与微生物互作研究亦有一定的意义。
1 材料与方法
1.1 植物材料
盆栽感黄矮病的小麦品系中8601 (每盆20株, 共2盆)在温室中(昼25℃, 14 h; 夜15℃, 10 h)。至二叶一心期, 在幼苗基部叶鞘处分别接种带BYDV-GAV或BYDV-PAV的蚜虫(中国农业科学院植物保护研究所王锡锋研究员和刘艳博士提供), 各20株。约35 d后开始显症, 取感病(含BYDV)叶片迅速放置液氮中速冻、-80℃保存。采用TRIzol (Invitrogen)方法提取叶片总RNA, 利用反转录试剂盒(TaKaRa DRR019)进行反转录获得第一链cDNA, 用于克隆BYDV-GAV、BYDV-PAV的CP完整ORF。
1.2 基因克隆
根据BYDV-GAV (GenBank登录号为AY220739.1), BYDV-PAV (EU332308.1)基因组中CP序列, 设计克隆BYDV-GAV和-PAV的 CP的引物GAV-CP-F、GAV-CP-R、PAV-CP-F和PAV-CP-R (表1), 以含BYDV的中8601 cDNA为扩增模板, 应用高保真酶进行PCR扩增, 1%琼脂糖电泳分离获得600 bp扩增产物。回收PCR产物, 将其产物插入克隆载体pMD18-T (TaKaRa D101), 转化至大肠杆菌TOP10感受态细胞中, 经过筛选、测序, 获得正确的阳性克隆pMD18-T-GAV-CP和pMD18-T-PAV-CP。 TiDPK1 (GenBank登录号为HQ423150)由本实验室克隆。
| 表1 本研究中所用的引物序列 Table 1 Primers used in this study |
1.3 酵母双杂交载体的构建
为构建与GAL4-BD融合的BYDV-GAV CP/ -PAV CP的酵母表达载体, 设计了在这2个完整编码序列两端分别引入 BamH I与 Pst I酶切位点的引物(表1), 以pMD18-T-GAV CP/-PAV-CP质粒为模板、使用高保真酶扩增、获得加入上述酶切位点的BYDV-GAV CP/ -PAV CP完整编码序列, 回收PCR扩增产物。利用 BamH I、 Pst I双酶切上述PCR回收产物和酵母表达载体pGBKT7 (Clontech)质粒, 用T4 ligase连接, 转化大肠杆菌TOP10感受态细胞, 经过筛选、测序, 获得正确的融合蛋白的酵母表达载体pGBKT7-GAV-CP和pGBKT7-PAV-CP。以同样的方法将 TiDPK1克隆到AD靶蛋白载体pGADT7 (Clontech), 构建 TiDPK1与GAL4-AD融合的酵母表达载体pGADT7-TiDPK1。
1.4 BiFC表达载体的构建
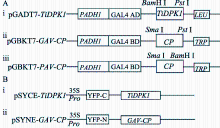
为将 TiDPK1和 BYDV-GAV CP完整的ORF分别融合到BIFC基础载体pSYCE和pSYNE载体中黄色荧光蛋白的C端一半、N端一半片段上, 利用Fast PCR Clone Kit (购买自PUEX公司)构建BiFC融合表达载体(图2)。设计引物扩增 TiDPK1和 GAV-CP完整的ORF, 将线性化载体 Sac I酶切位点两端邻近15 bp载体序列分别引入上、下游引物, 选择内切酶 Sac I分别消化骨架质粒 pSYCE、 pSYNE (中国科学院植物研究所林金星研究员惠赠), 将回收PCR产物、线性化载体按2∶1的比例混合, 加入FC重组酶, 室温反应30 min, 冰上放置5 min, 转化大肠杆菌TOP10感受态细胞, 经过筛选、测序, 获得正确的重组BiFC表达载体pSYCE-TiDPK1和pSYNE-GAV-CP。
1.5 酵母双杂交分析
YTH中所用酵母菌株为AH109, 含有 ADE2、 HIS3、 MEL1和 LacZ等报告基因, 这些报告基因表达受GAL4调控。只有在与GAL4-AD/-BD融合的2个蛋白互作、GAL4活性完整存在下, 酵母AH109细胞才能在-Ade/-His/-Leu/-Trp缺陷性培养基上正常生长, 能把无色底物X-α-gal水解为蓝色产物。
将AH109菌株在YPDA固体培养基上划线培养(30℃, 2~3 d), 挑选白色单克隆(直径2 mm左右)接种于1 mL YPDA液体培养基中, 30℃, 230 转 min-1培养8~12 h, 转接至50 mL (于250 mL三角瓶)继续振荡培养至OD600值为0.15~0.30。室温离心弃上清液, 将菌体重悬于100 mL YPDA液体培养基中培养直至OD600值为0.6。室温离心, 弃上清液, 将菌体重悬于60 mL无菌水。室温离心, 弃上清液, 将菌体重悬于3 ml 1.1倍的TE/LiAc中, 短暂离心, 弃上清液, 菌种重悬于600 μL 1.1×TE/溶液中, 获得了酵母感受态细胞, 立即用于转化实验。
提取pGADT7-TiDPK1、pGBKT7-GAV-CP、pGBKT7-PAV-CP等载体质粒、浓缩至1 μg μL-1以上, 采用PEG/LiAc法转化酵母感受态细胞[22]。pGADT7-TiDPK1、pGBKT7-GAV-CP与pGBKT7- PAV-CP单独转化, 用于毒理测试及自激活检验; pGADT7-TiDPK1+pGBKT7、pGADT7+pGBKT7-GAV- CP和pGADT7+pGBKT7-PAV-CP用于对照实验; pGADT7-TiDPK1+pGBKT7-GAV-CP和pGADT7- TiDPK1+pGBKT7-PAV-CP作为目标蛋白间互作分析的实验组。
1.6 BiFC分析
取新鲜的黄皮洋葱内第5或第6层表皮细胞约4~6 cm2, 内表皮朝下、紧贴于MS固体培养基中间位置, 避免产生气泡。密封后, 避光30℃培养4~5 h。提取pSYCE-TiDPK1和pSYNE-GAV-CP重组载体质粒, 各3 μg制作转化的金弹, 按照PDS 1000/He Particle Delivery System (Bio-Rad)手册进行基因枪转化上述培养的洋葱表皮细胞。同时转化4组不同组合的质粒, pSYCE-TiDPK1+pSYNE-GAV-CP、pSYCE+pSYNE、pSYCE+pSYNE-GAV-CP和pSYCE- TiDPK1+pSYNE, 每组组合中质粒按等量混合(各3 μg)。转化后的洋葱表皮细胞, 密封、避光30℃培养约16 h, 在共聚焦显微镜(Zeiss, LSM700) 513 nm波长下观察黄色荧光信号。
2 结果与分析
2.1 酵母双杂交重组载体及BiFC重组表达载体
以含BYDV的中8601 cDNA作为模板, 扩增获得BYDV -GAV CP和BYDV -PAV CP完整ORF, 大小为600 bp (图1)。序列比较结果表明, 与GenBank中注册的序列相比, BYDV- GAV CP序列完全一致, 而BYDV- PAV CP序列一致性为99%, 存在2个碱基的差异。
 | 图1 BYDV-GAV和BYDV-PAV外壳蛋白(CP)基因 开放阅读框的PCR扩增结果Fig. 1 PCR products of coat protein (CP) gene sequences of BYDV-GAV and BYDV-PAV1: BYDV-GAV CP; 2: BYDV-PAV CP; M: marker II. |
 | 图2 酵母双杂交系统(A)及BiFC系统(B)中 TiDPK1和BYDV CP的表达载体示意图Fig. 2 Scheme of TiDPK1 and CP expression vectors in YTH (A) and BiFC (B) |
将 TiDPK1和BYDV- GAV CP/- PAV CP的全长ORF分别构建到YTH载体pGADT7, pGBKT7中, 获得重组表达载体pGADT7-TiDPK和pGBKT7- GAV-CP/-PAV-CP (图2-A)。
将 TiDPK1和 GAV CP完整的ORF分别融合到BiFC基础载体pSYCE、pSYNE载体中黄色荧光蛋白的C端一半、N端一半上, 获得BiFC融合表达载体pSYCE-TiDPK1和pSYNE-GAV-CP (图2-B)。
2.2 酵母双杂交分析TiDPK1与GAV CP/-PAV CP间互作
将pGADT7-TiDPK1与pGBKT7-GAV-CP或pGBKT7-PAV-CP转化的酵母菌株分别涂布于SD/-Leu和SD/-Trp固体培养基, 30℃、培养2 d, 挑取单克隆于SD/-Leu/Amp+和SD/-Trp/Kan+液体培养基中进行毒理检测。30℃下230转 min-1培养过夜, 经检测, 培养基OD600值为0.923 (>0.8), 表明pGADT7-TiDPK1、pGBKT7-GAV-CP与pGBKT7- PAV-CP表达的融合蛋白对酵母细胞无毒副作用。
将pGADT7-TiDPK1和pGBKT7-GAV-CP/-PAV- CP转化的酵母菌株分别涂布于SD/-Leu和SD/-Trp固体平板, 30℃培养2 d, 挑取单克隆于0.5 mL YPD液体培养基中。30℃下230转min-1培养过夜, 稀释后分别涂布于营养缺陷性SD/-Leu/-His和SD/-Trp/ -His固体平板(添加30 mmol L-1 3AT, 3-amino-1,2, 4-triazole, 自激活抑制剂)。30℃倒置培养3 d, 均未出现生长细胞, 表明pGADT7-TiDPK1、pGBKT7- GAV-CP和pGBKT7-PAV-CP转化的酵母无自激活现象, 可用于后续实验。
将对照组和实验组重组质粒转化的酵母分别涂布于SD/-Leu/-Trp固体平板。30℃、培养3 d, 所有菌落正常生长。将上述单克隆的菌液涂布于含3AT的SD/-Ade/-His/-Leu/-Trp四缺营养缺陷性固体平板上(添加30 mmol L-1 3AT以增加可靠性。3AT是HIS3产物竞争抑制剂, 只有HIS3高表达酵母才能成活), 30℃下培养3 d后, pGADT7-TiDPK1+ pGBKT7-GAV-CP及pGADT7-TiDPK1+pGBKT7- PAV-CP转化的菌株开始出现菌落, 5 d后上述实验组的菌落生长良好(图3), 而pGADT7-TiDPK1+ pGBKT7、pGADT7+pGBKT7-GAV-CP和pGADT7+ pGBKT7-PAV-CP等对照菌株均未正常生长, 初步说明TiDPK1蛋白能够与BYDV-GAV、-PAV的CP互作。将对照组和实验组的酵母分别涂布于含X-α-gal的YPD固体培养基上, 结果对照组菌落无颜色变化, 而pGADT7-TiDPK1+pGBKT7-GAV-CP与pGADT7- TiDPK1+pGBKT7-PAV-CP实验组菌落变蓝(实验2 h后开始变蓝, 6 h后深蓝), 进一步表明TiDPK1在酵母双杂交系统内与BYDV-GAV和BYDV-PAV的CP具有相互作用(图3)。
 | 图3 酵母双杂交系统检验TiDPK1与BYGA-GAV CP/-PAV CP互作1: 转化实验组质粒(pGADT7-TiDPK1+pGBKT7-GAV-CP, pGADT7-TiDPK1+pGBKT7-PAV-CP)的酵母在四缺营养选择培养基上生长良好, 在X-α-gal定性实验变蓝; 2和3: 转化对照组质粒的酵母(pGADT7-TiDPK1+pGBKT7、pGADT7+pGBKT7-GAV-CP和pGADT7+pGBKT7-PAV-CP)不能在四缺培养基上生长, 同时X-α-gal定性实验无颜色变化。Fig. 3 BYDV-GAV CP/-PAV CP interacts with TiDPK1 in the YTH system1: Yeast colonies containing pGBKT7-CP/pGADT7-TiDPK1 grew well on SD/-Ade/-His/-Leu/-Trp medium and turned blue in X-α-gal assays; 2 and 3: Yeast colonies containing controls plasmids unable to grow on SD/-Ade/-His/-Leu/-Trp medium and turned blue in X-α-gal assays. |
2.3 BiFC分析TiDPK1与BYGA-GAV CP相互作用
利用基因枪将4种组合质粒(pSYCE+pSYNE、pSYCE+pSYNE-GAV-CP、pSYCE-TiDPK1+pSYNE、pSYCE-TiDPK1+pSYNE-GAV-CP)共转化至洋葱表皮细胞中, 瞬时表达。共聚焦荧光显微镜检测黄色荧光信号。结果表明, 共同转化pSYCE-TiDPK1+ pSYNE-GAV-CP的洋葱表皮细胞能表达YFP荧光 信号(图4-A), 而三组对照质粒的洋葱表皮细胞在共聚焦显微镜中检测不到YFP荧光信号(图4-B, -C, -D), 进一步证明TiDPK1与BYDV-GAV-CP能相互作用。
 | 图4 BiFC重组质粒共转化洋葱表皮细胞的共聚焦荧光及明场图像Fig. 4 Fluorescence and merged with bright-field images for the indicated proteins transiently expressing in onion epidermal cellsA: pSYCE-TiDPK1(CE-TiDPK1), pSYNE-GAV-CP(NE-CP); B: pSYCE-TiDPK1(CE-TiDPK1)+pSYNE(NE); C: pSYCE (CE)+pSYNE-GAV-CP(NE-CP); D: pSPYNE(NE)+pSPYCE(CE). |
3 讨论
植物在长期进化中形成精细的内在免疫系统, 以抵御入侵的病原微生物。植物通过细胞表面的pattern recognition receptors (PRRs)感知保守的病原相关分子(pattern-associated molecular patterns, PAMPs), 诱导产生初级的主动防御反应——表型激发免疫(PAMP-triggered immunity, PTI)。然而, 病原微生物又进化出抑制植物PTI的方法, 如通过分泌效应子(effector)到植物细胞内; 一旦病原获得抑制植物初级防御的能力, 植物即发展出第二层免疫反应—效应子激发免疫(effector-triggered immunity, ETI), 主要由 R基因编码蛋白产物直接识别或间接识别相应的病原微生物效应子, 激活防御反应信号、产生超敏反应和一系列防御反应物质, 最终产生抗病性[23]。蛋白激酶作为识别病原PAMPS的受体、辅助蛋白或传递信号的重要因子, 在植物抗病防御反应中具有重要作用[24]。
张增艳课题组发现TiDPK1蛋白激酶, 分布于细胞膜和细胞核中(待发表), 暗示其具备接收或传递病原信号的潜能。BYDV CP为黄矮病毒感染小麦的主要致病因子。因此, 研究TiDPK1与BYDV CP之间的关系, 对研究 TiDPK1的作用机制具有重要意义。YTH技术在研究植物抗病基因与病原物互作中被广泛应用。利用该技术已发现Pto与AvrPto[25]、RRS1-R全长蛋白与PopP2[26]及RIN4与AvrB或AvrRpm1[27]存在互作, RIN4与AvrB或AvrRpm1互作为RPM1介导的抗病反应所必需[27]。尽管YTH具有操作简单、结果明显等优点, 但在实验中难以彻底避免假阳性或假阴性结果, 因此相关结果常需用其他技术的实验验证。近年来, BiFC技术亦在植物与病原物互作研究中被广泛应用。同时使用YTH系统和BiFC系统, 能进一步降低假阳性, 为后续试验提高可靠的依据。Bar等[28]通过YTH及BiFC分析, 证明BAK1与LeEix1互作, 在烟草及马铃薯中促进了LeEix2对病原效应子EiX的反应; Burch-Smith等[29]通过BiFC、细胞生物化学和免疫共沉淀分析, 发现烟草抗病蛋白N与TMV病毒效应子p50互作, 并进一步指明N蛋白中的TIR亚结构域是二者互作所必需的。Wang等[30]通过YTH及BiFC等分析, 证明拟南芥抗病蛋白RPW8.2通过与PAPP2C互作, 进而防御白粉菌。本研究通过YTH证明TiDPK1能与BYDV-GAV-CP、BYDV-PAV-CP互作, 通过及BiFC系统进一步验证该实验结果。BYDV的CP是重要的致病因子, 本文通过YTH和BiFC实验证明TiDPK1可与BYDV CP互作, 为 TiDPK1抗BYDV作用机制提供重要依据。
4 结论
酵母双杂交实验结果和双分子荧光互补实验结果表明, TiDPK1蛋白激酶能与BYDV-GAV、-PAV的CP互作。
致谢: 感谢中国农业科学院植物保护研究所王锡锋研究员和刘艳博士提供带BYDV的蚜虫、中国科学院植物研究所林金星教授惠赠BiFC基础载体。
The authors have declared that no competing interests exist.
作者已声明无竞争性利益关系。
参考文献
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|