欢迎访问作物学报,今天是
作物学报 ›› 2021, Vol. 47 ›› Issue (11): 2121-2133.doi: 10.3724/SP.J.1006.2021.04249
曾健( ), 徐先超, 徐昱斐, 王秀成, 于海燕, 冯贝贝, 邢光南*()
), 徐先超, 徐昱斐, 王秀成, 于海燕, 冯贝贝, 邢光南*()
ZENG Jian(), XU Xian-Chao, XU Yu-Fei, WANG Xiu-Cheng, YU Hai-Yan, FENG Bei-Bei, XING Guang-Nan*()
摘要:
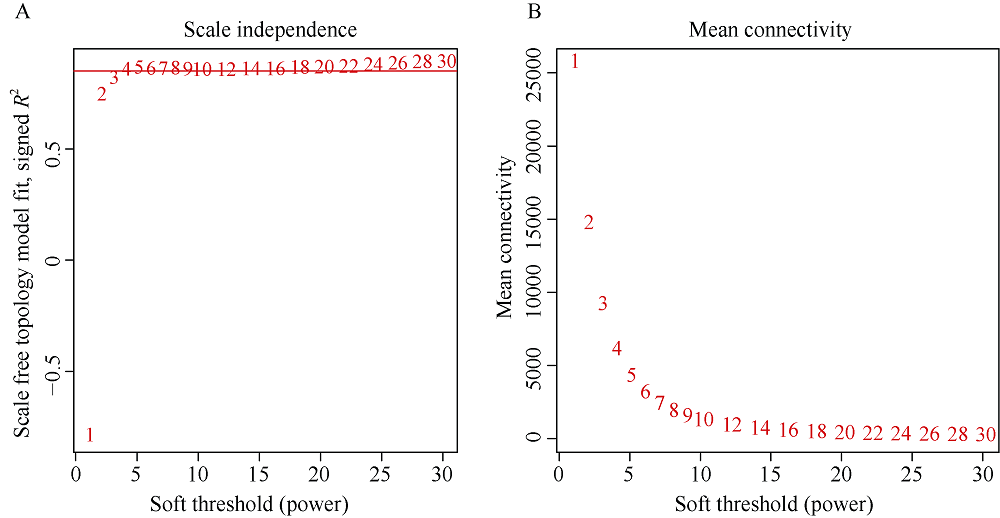
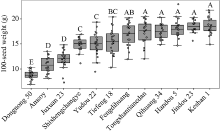
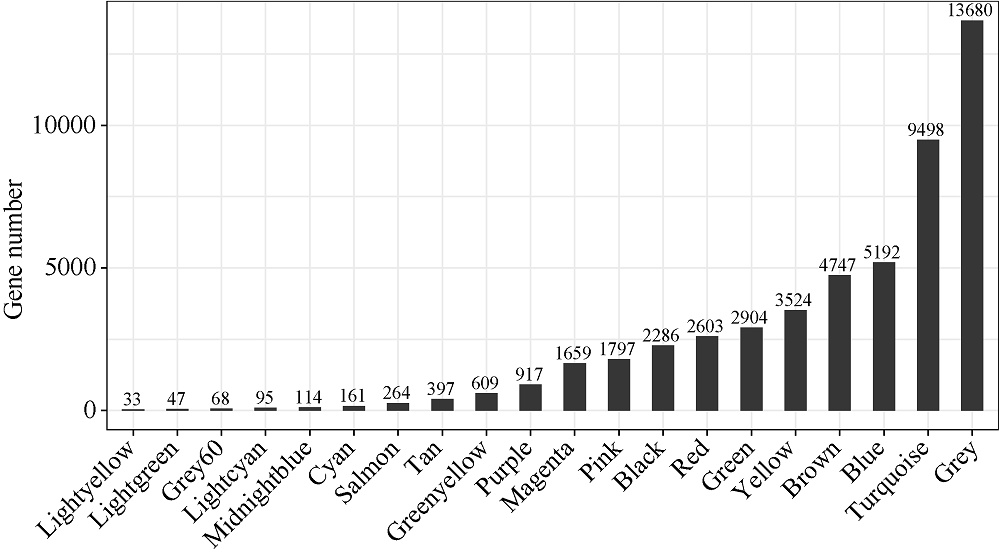
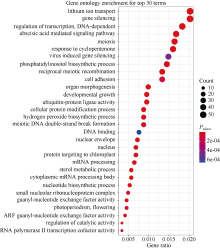
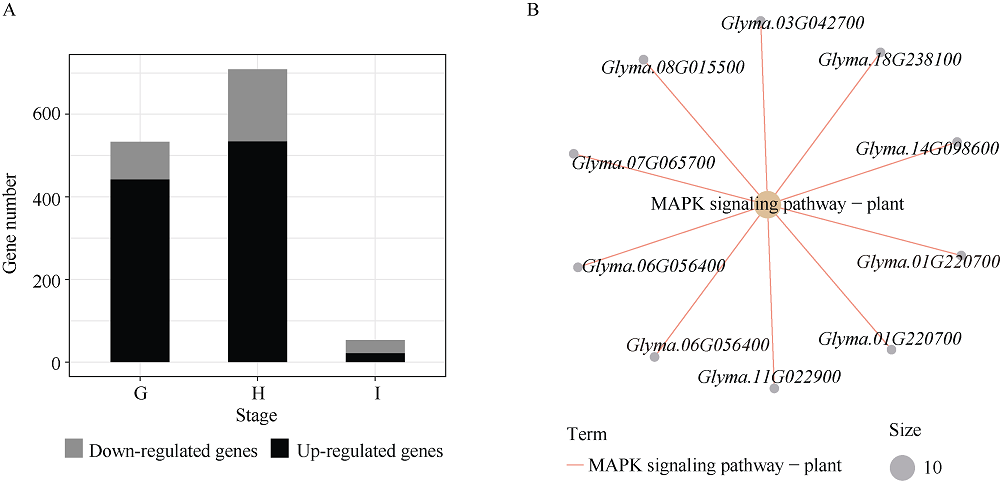
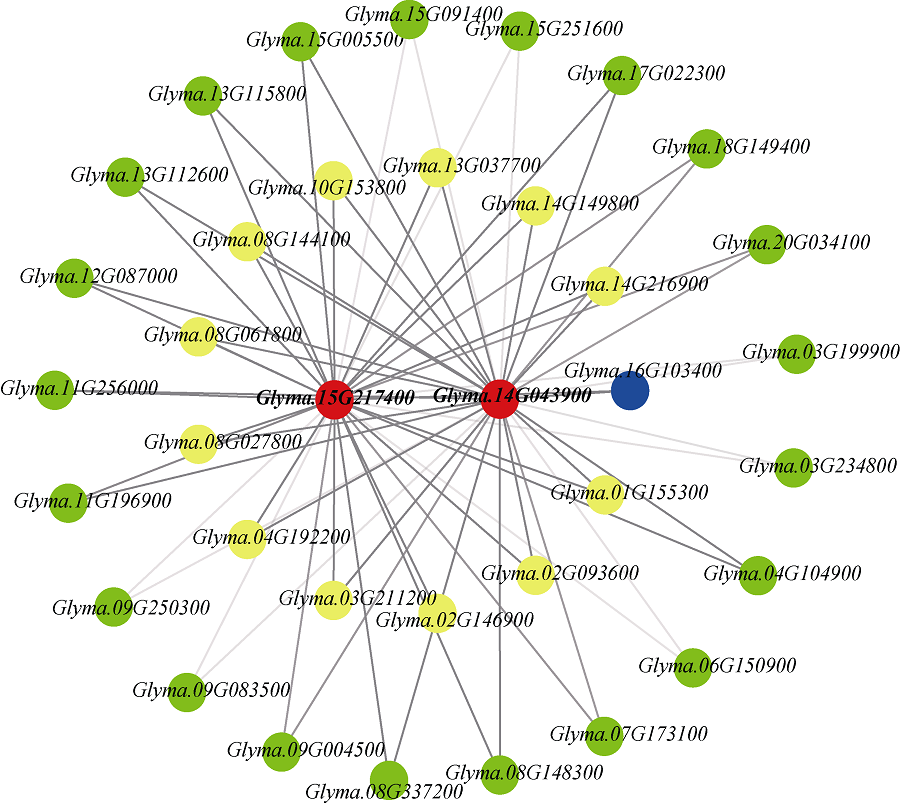
百粒重是影响大豆产量的重要农艺性状, 揭示其分子基础发掘关键候选基因对大豆改良具有重要意义。本研究通过对12个大豆品种籽粒发育3个时期共36个样本的转录组数据进行加权基因共表达网络分析(weighted gene co-expression network analysis, WGCNA), 得到20个基因共表达模块, 与百粒重及4个粒形性状关联后发现green模块与表型最为相关, 之后根据Gene Significance (GS)值和Eigengene Connectivity (kME)值筛选出13个green模块内的核心基因(hub gene); 然后对2组百粒重存在极显著差异的大豆品种的籽粒发育3个时期分别进行基因差异表达分析发现大豆在籽粒发育前中期可能通过MAPK信号通路调节百粒重大小; 之后对其进行SNPs/InDels挖掘并根据Gene Ontology (GO)注释发现green模块内的Glyma.14G043900和Glyma.15G217400由于SNP变异造成同义以及非同义突变, 且存在调控基因表达相关的GO Terms以及锌指结构域, 表明它们可能通过调控hub基因和差异表达基因调控大豆百粒重及粒形性状。Glyma.15G217400位于已报道的4个百粒重QTL中, 而Glyma.14G043900位于已报道的一个籽粒蛋白含量及一个油脂含量QTL中。通过比对大豆公共数据库发现这2个基因的百粒重增效等位变异受到人工选择, 其频率从野生大豆到地方品种再到育成品种的过程中逐渐升高。这些结果为进一步发掘大豆百粒重候选基因及其表达调控机制提供了新思路。
| [1] |
Lu X, Xiong Q, Cheng T, Li Q T, Liu X L, Bi Y D, Li W, Zhang W K, Ma B, Lai Y C, Du W G, Man W Q, Chen S Y, Zhang J S. A PP2C-1 allele underlying a quantitative trait locus enhances soybean 100-seed weight. Mol Plant, 2017, 10: 670-684.
doi: 10.1016/j.molp.2017.03.006 |
| [2] |
Li J, Zhao J, Li Y, Gao Y, Hua S, Nadeem M, Sun G, Zhang W, Hou J, Wang X, Qiu L. Identification of a novel seed size associated locus SW9-1 in soybean. Crop J, 2019, 7: 548-559.
doi: 10.1016/j.cj.2018.12.010 |
| [3] | 蒋费涛, 王书平, 祁俊生, 熊春霞. 转录组学技术及其在植物系统学上的研究进展. 现代盐化工, 2020, 47(4):14-17. |
| Jiang F T, Wang S P, Qi J S, Xiong C X. Research progress of transcriptional technology and its advances in plant phylogeny. Mod Salt Chem Ind, 2020, 47(4):14-17 (in Chinese with English abstract). | |
| [4] |
Rory S, Marta G, James H. RNA sequencing: the teenage years. Nat Rev Genet, 2019, 20: 631-656.
doi: 10.1038/s41576-019-0150-2 |
| [5] | 杨宇昕, 桑志勤, 许诚, 代文双, 邹枨. 利用WGCNA进行玉米花期基因共表达模块鉴定. 作物学报, 2019, 45: 161-174. |
| Yang Y X, Sang Z Q, Xu C, Dai W S, Zou C. Identification of co-expression modules for maize flowering stage genes using WGCNA. Acta Agron Sin, 2019, 45: 161-174 (in Chinese with English abstract). | |
| [6] |
Hollender C A, Kang C, Darwish O, Geretz A, Matthews B F, Slovin J, Alkharouf N, Liu Z. Floral transcriptomes in woodland strawberry uncover developing receptacle and anther gene networks. Plant Physiol, 2014, 165: 1062-1075.
pmid: 24828307 |
| [7] |
Greenham K, Guadagno C R, Gehan M A, Mockler T C, Weinig C, Ewers B E, McClung C R. Temporal network analysis identifies early physiological and transcriptomic indicators of mild drought in Brassica rapa. eLife, 2017, 6: e29655.
doi: 10.7554/eLife.29655 |
| [8] |
Yang S N, Miao L, He J B, Zhang K, Li Y, Gai J Y. Dynamic transcriptome changes related to oil accumulation in developing soybean seeds. Int J Mol Sci, 2019, 20: 2202.
doi: 10.3390/ijms20092202 |
| [9] |
Lu X, Li Q T, Xiong Q, Li W, Bi Y D, Lai Y C, Liu X L, Man W Q, Zhang W K, Ma B, Chen S Y, Zhang J S. The transcriptomic signature of developing soybean seeds reveals the genetic basis of seed trait adaptation during domestication. Plant J, 2016, 86: 530-544.
doi: 10.1111/tpj.2016.86.issue-6 |
| [10] | Lopez-Maestre H, Brinza L, Marchet C, Kielbassa J, Bastien S, Boutigny M, Monnin D, Filali A E, Carareto C M, Vieira C, Picard F, Kremer N, Vavre F, Sagot M F, Lacroix V. SNP calling from RNA-seq data without a reference genome: identification, quantification, differential analysis and impact on the protein sequence. Nucleic Acids Res, 2016, 44: e148. |
| [11] |
Rogier O, Chateigner A, Amanzougarene S, Lesage-Descauses M C, Balzergue S, Brunaud V, Caius J, Soubigou-Taconnat L, Jorge V, Segura V. Accuracy of RNAseq based SNP discovery and genotyping in Populus nigra. BMC Genomics, 2018, 19: 909.
doi: 10.1186/s12864-018-5239-z pmid: 30541448 |
| [12] |
Liu Y C, Du H L, Li P C, Shen Y T, Peng H, Liu S L, Zhou G A, Zhang H K, Liu Z, Shi M, Huang X H, Li Y, Zhang M, Wang Z, Zhu B G, Han B, Liang C Z, Tian Z X. Pan-genome of wild and cultivated soybeans. Cell, 2020, 182: 162-176.
doi: 10.1016/j.cell.2020.05.023 |
| [13] | 丁琦, 徐伟, 李蒙, 王秀成, 卢伟, 盖钧镒, 王玲, 邢光南. 基于分水岭和统计矩的大豆籽粒形态参数测量方法. 大豆科学, 2019, 38: 960-967. |
| Ding Q, Xu W, Li M, Wang X C, Lu W, Gai J Y, Wang L, Xing G N. Measurement method of soybean seed morphological parameters based on watershed and statistical moment. Soybean Sci, 2019, 38: 960-967 (in Chinese with English abstract). | |
| [14] |
Pertea M, Kim D, Pertea G M, Leek J T, Salzberg S L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc, 2016, 11: 1650-1667.
doi: 10.1038/nprot.2016.095 |
| [15] |
Michael I L, Wolfgang H, Simon A. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol, 2014, 15: 550.
pmid: 25516281 |
| [16] |
Peter L, Steve H. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics, 2008, 9: 1-32.
doi: 10.1186/1471-2105-9-1 |
| [17] |
Yu G C, Wang L G, Han Y Y, He Q Y. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics, 2012, 16: 284-287.
doi: 10.1089/omi.2011.0118 |
| [18] |
Paul S, Andrew M, Owen O, Nitin S B, Jonathan T W, Daniel R, Nada A, Benno S, Trey I. Cytoscape: a software environment for integrated. models of biomolecular interaction networks. Genome Res, 2003, 13: 2498-2504.
doi: 10.1101/gr.1239303 |
| [19] |
Engström P G, Steijger T, Sipos B, Grant G R, Kahles A, Rätsch G, Goldman N, Hubbard T J, Harrow J, Guigó R, Bertone P. Systematic evaluation of spliced alignment programs for RNA-seq data. Nat Methods, 2013, 10: 1185-1191.
doi: 10.1038/nmeth.2722 pmid: 24185836 |
| [20] |
Alexander D, Carrie A D, Felix S, Jorg D, Chris Z, Sonali J, Philippe B, Mark C, Thomas R G. STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 2013, 29: 15-21.
doi: 10.1093/bioinformatics/bts635 pmid: 23104886 |
| [21] |
Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics, 2011, 27: 2987-2993.
doi: 10.1093/bioinformatics/btr509 |
| [22] |
Heldenbrand J R, Baheti S, Bockol M A, Drucker T M, Hart S N, Hudson M E, Iyer R K, Kalmbach M T, Kendig K I, Klee E W, Mattson N R, Wieben E D, Wiepert M, Wildman D E, Mainzer L S. Recommendations for performance optimizations when using GATK3.8 and GATK4. BMC Bioinf, 2019, 20: 722.
doi: 10.1186/s12859-019-3277-4 |
| [23] |
Petr D, Adam A, Goncalo A, Cornelis A A, Eric B, Mark A D, Robert E H, Gerton L, Gabor T M, Stephen T S, Gilean M, Richard D. The variant call format and VCFtools. Bioinformatics, 2011, 27: 2156-2158.
doi: 10.1093/bioinformatics/btr330 |
| [24] |
Cingolani P, Platts A, Wang L L, Coon M, Nguyen T, Wang L, Land S J, Lu X Y, Ruden D M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly, 2012, 6: 80-92.
doi: 10.4161/fly.19695 pmid: 22728672 |
| [25] |
Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones S J, Marra M A. Circos: an information aesthetic for comparative genomics. Genome Res, 2009, 19: 1639-1645.
doi: 10.1101/gr.092759.109 pmid: 19541911 |
| [26] |
Torkamaneh D, Laroche J, Valliyodan B, O’Donoughue L, Belzile F. Soybean (Glycine max) Haplotype Map (GmHapMap): a universal resource for soybean translational and functional genomics. Plant Biotechnol J, 2020, 19: 324-334.
doi: 10.1111/pbi.v19.2 |
| [27] |
Li N, Xu R, Li Y. Molecular networks of seed size control in plants. Annu Rev Plant Biol, 2019, 70: 435-463.
doi: 10.1146/annurev-arplant-050718-095851 |
| [28] |
Cheng Z J, Zhao X Y, Shao X X, Wang F, Zhou C, Liu Y G, Zhang Y, Zhang X S. Abscisic acid regulates early seed development in Arabidopsis by ABI5-mediated transcription of SHORT HYPOCOTYL UNDER BLUE1. Plant Cell, 2014, 26: 1053-1068.
doi: 10.1105/tpc.113.121566 |
| [29] |
Koichiro A, Tokunori H, Kanna S I, Miyako U T, Hidemi K, Makoto M. A novel AP2-type transcription factor, SMALL ORGAN SIZE1, controls organ size downstream of an auxin signaling pathway. Plant Cell Physiol, 2014, 55: 897-912.
doi: 10.1093/pcp/pcu023 pmid: 24486766 |
| [30] |
Gu Y Z, Li W, Jiang H W, Wang Y, Gao H H, Liu M, Chen Q S, Lai Y C, He C Y. Differential expression of a WRKY gene between wild and cultivated soybeans correlates to seed size. J Exp Bot, 2017, 68: 2717-2729.
doi: 10.1093/jxb/erx147 |
| [31] |
Li N, Li Y H. Signaling pathways of seed size control in plants. Curr Opin Plant Biol, 2016, 33: 23-32.
doi: 10.1016/j.pbi.2016.05.008 |
| [32] |
Hans W, Ljudmilla B, Ulrich W. Molecular physiology of legume seed development. Annu Rev Plant Biol, 2005, 56: 253-279.
doi: 10.1146/annurev.arplant.56.032604.144201 |
| [33] | 倪资园, 蔡丽, 谢皓, 陈学珍. 大豆籽粒发育过程中营养物质的积累动态分析. 北京农学院学报, 2011, 26(2):1-3. |
| Ni Z Y, Cai L, Xie H, Chen X Z. Analysis of dynamic accumulation on nutrients during maturing process in soybean seed. J Beijing Agric Coll, 2011, 26(2):1-3 (in Chinese with English abstract). | |
| [34] |
Xu R, Duan P G, Yu H Y, Zhou Z K, Zhang B L, Wang R C, Li J, Zhang G Z, Zhuang S S, Lyu J, Li N, Chai T Y, Tian Z X, Yao S G, Li Y H. Control of grain size and weight by the OsMKKK10-OsMKK4-OsMAPK6 signaling pathway in rice. Mol Plant, 2018, 11: 860-873.
doi: 10.1016/j.molp.2018.04.004 |
| [35] |
Srivastava A K, Lu Y M, Zinta G, Lang Z B, Zhu J K. UTR-dependent control of gene expression in plants. Trends Plant Sci, 2018, 23: 248-259.
doi: S1360-1385(17)30255-8 pmid: 29223924 |
| [36] |
Mian M A, Bailey M A, Tamulonis J P, Shipe E R, Carter T E, Parrott W A, Ashley D A, Hussey R S, Boerma H R. Molecular markers associated with seed weight in two soybean populations. Theor Appl Genet, 1996, 93: 1011-1016.
doi: 10.1007/BF00230118 pmid: 24162474 |
| [37] |
Orf J H, Chase K, Jarvik T, Mansur L M, Cregan P B, Adler F R, Lark K G. Genetics of soybean agronomic traits: I. Comparison of three related recombinant inbred populations. Crop Sci, 1999, 39: 1642-1651.
doi: 10.2135/cropsci1999.3961642x |
| [38] | Li W X, Zheng D H. QTL mapping for major agronomic traits across two years in soybean (Glycine max L. Merr.). J Crop Sci Biotechnol, 2008, 11: 171-176. |
| [39] |
Teng W, Han Y, Du Y, Sun D, Zhang Z, Qiu L, Sun G, Li W. QTL analyses of seed weight during the development of soybean (Glycine max L. Merr.). Heredity, 2009, 102: 372-380.
doi: 10.1038/hdy.2008.108 pmid: 18971958 |
| [40] |
Akond M, Liu S M, Boney M, Kantartzi St K, Meksem K, Bellaloui N, Lightfoot D A, Kassem M A. Identification of quantitative trait loci (QTL) underlying protein, oil, and five major fatty acids’ contents in soybean. Am J Plant Sci, 2014, 5: 158-167.
doi: 10.4236/ajps.2014.51021 |
| [1] | 陈玲玲, 李战, 刘亭萱, 谷勇哲, 宋健, 王俊, 邱丽娟. 基于783份大豆种质资源的叶柄夹角全基因组关联分析[J]. 作物学报, 2022, 48(6): 1333-1345. |
| [2] | 田甜, 陈丽娟, 何华勤. 基于Meta-QTL和RNA-seq的整合分析挖掘水稻抗稻瘟病候选基因[J]. 作物学报, 2022, 48(6): 1372-1388. |
| [3] | 杨欢, 周颖, 陈平, 杜青, 郑本川, 蒲甜, 温晶, 杨文钰, 雍太文. 玉米-豆科作物带状间套作对养分吸收利用及产量优势的影响[J]. 作物学报, 2022, 48(6): 1476-1487. |
| [4] | 王炫栋, 杨孙玉悦, 高润杰, 余俊杰, 郑丹沛, 倪峰, 蒋冬花. 拮抗大豆斑疹病菌放线菌菌株的筛选和促生作用及防效研究[J]. 作物学报, 2022, 48(6): 1546-1557. |
| [5] | 于春淼, 张勇, 王好让, 杨兴勇, 董全中, 薛红, 张明明, 李微微, 王磊, 胡凯凤, 谷勇哲, 邱丽娟. 栽培大豆×半野生大豆高密度遗传图谱构建及株高QTL定位[J]. 作物学报, 2022, 48(5): 1091-1102. |
| [6] | 李阿立, 冯雅楠, 李萍, 张东升, 宗毓铮, 林文, 郝兴宇. 大豆叶片响应CO2浓度升高、干旱及其交互作用的转录组分析[J]. 作物学报, 2022, 48(5): 1103-1118. |
| [7] | 彭西红, 陈平, 杜青, 杨雪丽, 任俊波, 郑本川, 罗凯, 谢琛, 雷鹿, 雍太文, 杨文钰. 减量施氮对带状套作大豆土壤通气环境及结瘤固氮的影响[J]. 作物学报, 2022, 48(5): 1199-1209. |
| [8] | 王好让, 张勇, 于春淼, 董全中, 李微微, 胡凯凤, 张明明, 薛红, 杨梦平, 宋继玲, 王磊, 杨兴勇, 邱丽娟. 大豆突变体ygl2黄绿叶基因的精细定位[J]. 作物学报, 2022, 48(4): 791-800. |
| [9] | 李瑞东, 尹阳阳, 宋雯雯, 武婷婷, 孙石, 韩天富, 徐彩龙, 吴存祥, 胡水秀. 增密对不同分枝类型大豆品种同化物积累和产量的影响[J]. 作物学报, 2022, 48(4): 942-951. |
| [10] | 杜浩, 程玉汉, 李泰, 侯智红, 黎永力, 南海洋, 董利东, 刘宝辉, 程群. 利用Ln位点进行分子设计提高大豆单荚粒数[J]. 作物学报, 2022, 48(3): 565-571. |
| [11] | 周悦, 赵志华, 张宏宁, 孔佑宾. 大豆紫色酸性磷酸酶基因GmPAP14启动子克隆与功能分析[J]. 作物学报, 2022, 48(3): 590-596. |
| [12] | 王娟, 张彦威, 焦铸锦, 刘盼盼, 常玮. 利用PyBSASeq算法挖掘大豆百粒重相关位点与候选基因[J]. 作物学报, 2022, 48(3): 635-643. |
| [13] | 董衍坤, 黄定全, 高震, 陈栩. 大豆PIN-Like (PILS)基因家族的鉴定、表达分析及在根瘤共生固氮过程中的功能[J]. 作物学报, 2022, 48(2): 353-366. |
| [14] | 张国伟, 李凯, 李思嘉, 王晓婧, 杨长琴, 刘瑞显. 减库对大豆叶片碳代谢的影响[J]. 作物学报, 2022, 48(2): 529-537. |
| [15] | 禹桃兵, 石琪晗, 年海, 连腾祥. 涝害对不同大豆品种根际微生物群落结构特征的影响[J]. 作物学报, 2021, 47(9): 1690-1702. |
|
||