欢迎访问作物学报,今天是
作物学报 ›› 2023, Vol. 49 ›› Issue (9): 2321-2330.doi: 10.3724/SP.J.1006.2023.23072
• 作物遗传育种·种质资源·分子遗传学 • 下一篇
杨文宇1,2( ), 吴成秀1, 肖英杰1,3,*(), 严建兵1,3
), 吴成秀1, 肖英杰1,3,*(), 严建兵1,3
YANG Wen-Yu1,2(), WU Cheng-Xiu1, XIAO Ying-Jie1,3,*(), YAN Jian-Bing1,3
摘要:
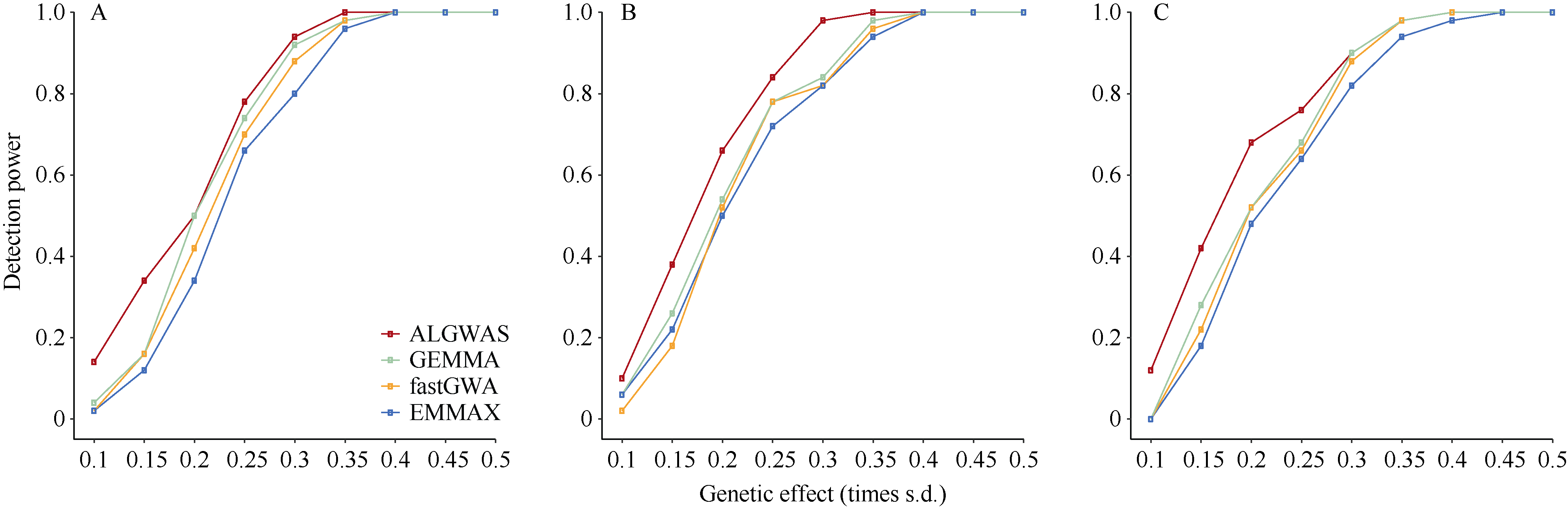
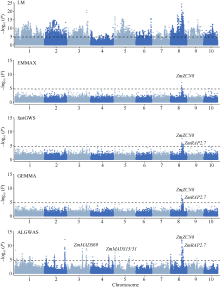
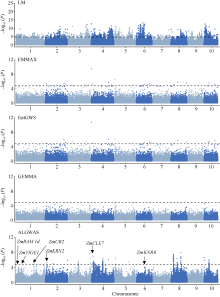
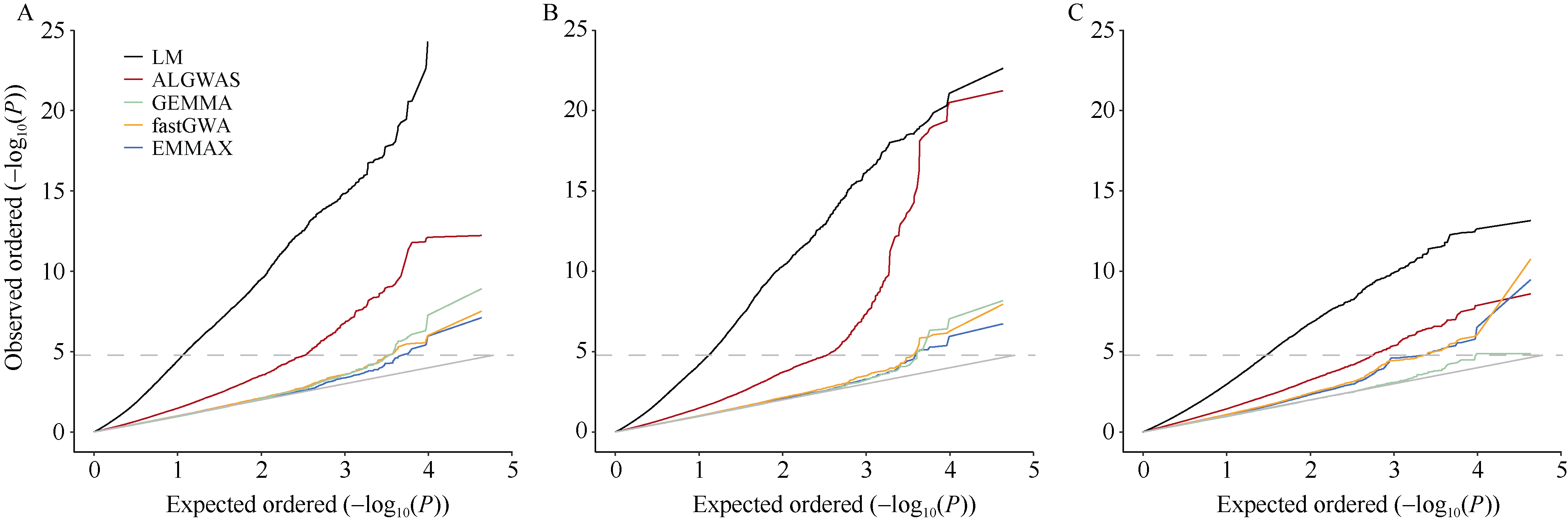
作为进行全基因组关联分析的主流方法, 混合线性模型类方法得到了广泛的应用。但是, 现有方法仍存在检测功效不高的问题。本文提出一种基于Adaptive Lasso的2阶段全基因组关联分析方法(two-stage Adaptive Lasso-based genome-wide association analysis, ALGWAS), 该方法在第1阶段通过变量选择方法Adaptive Lasso筛选出与目标性状相关联的单核苷酸多态性位点(single nucleotide polymorphism, SNP), 第2阶段将第1阶段筛选出的SNP作为协变量放入线性模型中进行全基因组扫描。在模拟实验中, ALGWAS方法与3种常用的全基因组关联分析方法fastGWA、GEMMA和EMMAX相比具有最高的检测功效, 同时具有较低的错误发现率(false discovery rate, FDR)。将以上4种方法应用到包含1341份材料的玉米CUBIC (Complete-diallel plus Unbalanced Breeding-like Inter-Cross)群体的全基因组关联分析中, ALGWAS方法可检测到与开花期相关基因ZmMADS69、ZmMADS15/31、ZmZCN8和ZmRAP2.7, 与株高相关基因ZmBRD1和ZmBR2, 与产量相关基因ZmUB2、ZmKRN2和ZmCLE7等, 而其他3种常用的全基因组关联分析方法检测功效较低。本研究提出了一种非混合线性模型类的全基因组关联分析方法, 对解析微效多基因决定的复杂遗传性状具有更高的检测效率, 为基因挖掘提供了新的途径。
| [1] |
Zhang Y M, Mao Y C, Xie C Q, Smith H, Luo L, Xu S Z. Mapping quantitative trait loci using naturally occurring genetic variance among commercial inbred lines of maize (Zea mays L.). Genetics, 2005, 169: 2267-2275.
doi: 10.1534/genetics.104.033217 |
| [2] |
Yu J M, Pressoir G, Briggs H W, Vroh B I, Yamasakiet M, Doebley J F, McMullen M D, Gaut B S, Nielsen D M, Holland J B, Kresovich S, Buckler E S. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet, 2006, 38: 203-208.
doi: 10.1038/ng1702 pmid: 16380716 |
| [3] |
Kang H M, Zaitlen N A, Wade C M, Kirby A, Heckerman D, Daly M J, Eskin E. Efficient control of population structure in model organism association mapping. Genetics, 2008, 178: 1709-1723.
doi: 10.1534/genetics.107.080101 pmid: 18385116 |
| [4] |
Kang H M, Sul J H, Service S K, Zaitlen N A, Kong S Y, Freimer N B, Sabatti C, Eskin E. Variance component model to account for sample structure in genome-wide association studies. Nat Genet, 2010, 42: 348-354.
doi: 10.1038/ng.548 pmid: 20208533 |
| [5] |
Zhang Z W, Ersoz E, Lai C Q, Todhunter R J, Tiwari H K, Gore M A, Bradbury P J, Yu J, Arnett D K, Ordovas J M, Buckler E S. Mixed linear model approach adapted for genome-wide association studies. Nat Genet, 2010, 42: 355-360.
doi: 10.1038/ng.546 pmid: 20208535 |
| [6] |
Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat Genet, 2012, 44: 821-824.
doi: 10.1038/ng.2310 pmid: 22706312 |
| [7] |
Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3000 shared controls. Nature, 2007, 447: 661-678.
doi: 10.1038/nature05911 |
| [8] |
Li H, Peng Z Y, Yang X H, Wang W D, Fu J J, Wang J H, Han Y J, Chai Y C, Guo T T, Yang N, Liu J, Warburton M L, Cheng Y B, Hao X M, Zhang P, Zhao J Y, Liu Y J, Wang G Y, Li J S, Yan J B. Genome-wide association study dissects the genetic architecture of oil biosynthesis in maize kernels. Nat Genet, 2013, 45: 43-50.
doi: 10.1038/ng.2484 pmid: 23242369 |
| [9] |
Huang X H, Wei X H, Sang T, Zhao Q, Feng Q, Zhao Y, Li C Y, Zhu C R, Lu T T, Zhang Z W, Li M, Fan D L, Guo Y L, Wang A, Wang L, Deng L W, Li W J, Lu Y Q, Weng Q J, Liu K Y, Huang T, Zhou T Y, Jing Y F, Li W, Lin Z, Buckler E S, Qian Q, Zhang Q F, Li J Y, Han B. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat Genet, 2010, 42: 961-969.
doi: 10.1038/ng.695 pmid: 20972439 |
| [10] |
Xiao Y J, Liu H J, Wu L J, Warburton M L, Yan J B. Genome- wide association studies in maize: praise and stargaze. Mol Plant, 2017, 10: 359-374.
doi: 10.1016/j.molp.2016.12.008 |
| [11] |
彭勃, 赵晓雷, 王奕, 袁文娅, 李春辉, 李永祥, 张登峰, 石云素, 宋燕春, 王天宇, 黎裕. 玉米叶向值的全基因组关联分析. 作物学报, 2020, 46: 819-831.
doi: 10.3724/SP.J.1006.2020.93063 |
|
Peng B, Zhao X L, Wang Y, Yuan W Y, Li C H, Li Y X, Zhang D F, Shi Y S, Song Y C, Wang T Y, Li Y. Genome-wide association studies of leaf orientation value in maize. Acta Agron Sin, 2020, 46: 819-831. (in Chinese with English abstract)
doi: 10.3724/SP.J.1006.2020.93063 |
|
| [12] |
谢磊, 任毅, 张新忠, 王继庆, 张志辉, 石书兵, 耿洪伟. 小麦穗发芽性状的全基因组关联分析. 作物学报, 2021, 47: 1891-1902.
doi: 10.3724/SP.J.1006.2021.01078 |
|
Xie L, Ren Y, Zhang X Z, Wang J Q, Zhang Z H, Shi S B, Geng H W. Genome-wide association study of pre-harvest sprouting traits in wheat. Acta Agron Sin, 2021, 47: 1891-1902. (in Chinese with English abstract)
doi: 10.3724/SP.J.1006.2021.01078 |
|
| [13] |
杨飞, 张征锋, 南波, 肖本泽. 水稻产量相关性状的全基因组关联分析及候选基因筛选. 作物学报, 2022, 48: 1813-1821.
doi: 10.3724/SP.J.1006.2022.12047 |
|
Yang F, Zhang Z F, Nan B, Xiao B Z. Genome-wide association analysis and candidate gene selection of yield related traits in rice. Acta Agron Sin, 2022, 48: 1813-1821. (in Chinese with English abstract)
doi: 10.3724/SP.J.1006.2022.12047 |
|
| [14] |
Lippert C, Listgarten J, Liu Y, Kadiel C M, Davidson R I, Heckerman D. FaST linear mixed models for genome-wide association studies. Nat Methods, 2011, 8: 833-835.
doi: 10.1038/nmeth.1681 pmid: 21892150 |
| [15] |
Listgarten J, Lippert C, Kadie C M, Davidson R I, Eskin E, Heckerman D. Improved linear mixed models for genome-wide association studies. Nat Methods, 2012, 9: 525-526.
doi: 10.1038/nmeth.2037 pmid: 22669648 |
| [16] |
Loh P R, Bhatia G, Gusev A, Finucane H K, Bulik-Sullivan B K, Pollack S J. Contrasting genetic architectures of schizophrenia and other complex diseases using fast variance-components analysis. Nat Genet, 2015, 47: 1385-1392.
doi: 10.1038/ng.3431 pmid: 26523775 |
| [17] |
Jiang L D, Zheng Z L, Qi T, Kemper K E, Wray N R, Visscher P M, Yang J. A resource-efficient tool for mixed model association analysis of large-scale data. Nat Genet, 2019, 51: 1749-1755.
doi: 10.1038/s41588-019-0530-8 pmid: 31768069 |
| [18] | Maher B. Personal genomes: the case of the missing heritability. Nature, 2008, 456: 18-21. |
| [19] |
Visscher P. Sizing up human height variation. Nat Genet, 2008, 40: 489-490.
doi: 10.1038/ng0508-489 pmid: 18443579 |
| [20] |
Yang J, Benyamin B, McEvoy B P, Gordon S, Henders A K, Nyholt D R, Madden P A, Heath A C, Martin N G, Montgomery G W, Goddard M E, Visscher P M. Common SNPs explain a large proportion of the heritability for human height. Nat Genet, 2010, 42: 565-569.
doi: 10.1038/ng.608 pmid: 20562875 |
| [21] | Song B, Mott R, Gan X. Recovery of novel association loci in Arabidopsis thaliana and Drosophila melanogaster through leveraging INDELs association and integrated burden test. PLoS Genet, 2018, 14: e1007699. |
| [22] | Zhang Y W, Tamba C L, Wen Y J, Li P, Ren W L, Ni Y L, Gao J, Zhang Y M. mrMLM v4.0.2: an R platform for multi-locus genome-wide association studies. Genom Prot Bioinfor, 2020, 18: 481-487. |
| [23] | Yang N, Lu Y L, Yang X H, Huang J, Zhou Y, Ali F H, Wen W W, Liu J, Li J S, Yan J B. Genome wide association studies using a new nonparametric model reveal the genetic architecture of 17 agronomic traits in an enlarged maize association panel. PLoS Genet, 2014, 10: e1004573. |
| [24] |
Liu H J, Wang X Q, Xiao Y J, Luo J Y, Qiao F, Yang W Y, Zhang R Y, Meng Y J, Sun J M, Yan S J, Peng Y, Niu L Y, Jian L M, Song W, Yan J L, Li C H, Zhao Y X, Liu Y, Warburton M L, Zhao J R, Yan J B. CUBIC: an atlas of genetic architecture promises directed maize improvement. Genome Biol, 2020, 21: 20.
doi: 10.1186/s13059-020-1930-x |
| [25] |
Lande R, Thompson R. Efficiency of marker-assisted selection in the improvement of quantitative traits. Genetics, 1990, 124: 743-756.
doi: 10.1093/genetics/124.3.743 pmid: 1968875 |
| [26] |
Yu J M, Holland J B, McMullen M D, Buckler E S. Genetic design and statistical power of nested association mapping in maize. Genetics, 2008, 178: 539-551.
doi: 10.1534/genetics.107.074245 pmid: 18202393 |
| [27] | Tibshirani R. Regression shrinkage and selection via the lasso. J Royal Statist Society, 1996, 58: 267-288. |
| [28] |
Zou H. The adaptive lasso and its oracle properties. J Am Statist Assoc, 2006, 101: 1418-1429.
doi: 10.1198/016214506000000735 |
| [29] |
Liang Y M, Liu Q, Wang X F, Huang C, Xu G H, Hey S, Lin H Y, Li C, Xu D Y, Wu L S, Wang C L, Wu W H, Xia J L, Han X, Lu S J, Lai J S, Song W B, Schnable P S, Tian F. ZmMADS69 functions as a flowering activator through the regulatory module and contributes to maize flowering time adaptation. New Phytol, 2019, 221: 2335-2347.
doi: 10.1111/nph.2019.221.issue-4 |
| [30] | Makarevitch I, Thompson A, Muehlbauer G J, Springer N M. Brd1 gene in maize encodes a brassinosteroid C-6 oxidase. PLoS One, 2012, 7: e30798. |
| [31] |
Xing A Q, Gao Y F, Ye L F, Zhang W P, Cai L C, Ching A, Llaca V, Johnson B, Liu L, Yang X H, Kang D M, Yan J B, Li J S. A rare SNP mutation in Brachytic2 moderately reduces plant height and increases yield potential in maize. J Exp Bot, 2015, 66: 3791-3802.
doi: 10.1093/jxb/erv182 pmid: 25922491 |
| [32] |
Yang N, Liu J, Gao Q, Gui S T, Chen L, Yang L F, Huang J, Deng T Q, Luo J Y, He L J, Wang Y B, Xu P W, Peng Y, Shi Z, Lan L, Ma Z Y, Yang X, Zhang Q Q, Bai M Z, Li W, Liu L, Jackson D, Yan J B. Genome assembly of a tropical maize inbred line provides insights into structural variation and crop improvement. Nat Genet, 2019, 51: 1052-1059.
doi: 10.1038/s41588-019-0427-6 pmid: 31152161 |
| [33] |
Luo Y, Zhang M L, Liu Y, Liu J, Li W Q, Chen G S, Peng Y, Jin M, Wei W J, Jian L M, Yan J, Fernie A R, Yan J B. Genetic variation in YIGE1 contributes to ear length and grain yield in maize. New Phytol, 2022, 234: 513-526.
doi: 10.1111/nph.v234.2 |
| [34] | Du Y F, Liu L, Peng Y, Li M F, Li Y F, Liu D, Li X W, Zhang Z X. UNBRANCHED3 expression and inflorescence development is mediated by UNBRANCHED2 and the distal enhancer, KRN4, in maize. PLoS Genet, 2020, 16: e1008764. |
| [35] | Chen W K, Chen L, Zhang X, Yang N, Guo J H, Wang M, Ji S G, Zhao X Y, Yin P F, Cai L C, Xu J, Zhang L L, Han Y J, Xiao Y N, Xu G, Wang Y B, Wang S H, Wu S, Yang F, Jackson D, Cheng J K, Chen S H, Sun C Q, Qin F, Tian F, Fernie A R, Li J S, Yan J B, Yang X H. Convergent selection of a WD40 protein that enhances grain yield in maize and rice. Science, 2022, 375: e7985. |
| [36] |
Liu L, Gallagher J, Arevalo E D, Chen R, Skopelitis T, Wu Q, Bartlett M, Jackson D. Enhancing grain-yield-related traits by CRISPR-Cas9 promoter editing of maize CLE genes. Nat Plants, 2021, 7: 287-294.
doi: 10.1038/s41477-021-00858-5 pmid: 33619356 |
| [37] |
Jia H T, Li M F, Li W Y, Liu L, Jian Y N, Yang Z X, Shen X M, Ning Q, Du Y F, Zhao R, Jackson D, Yang X H, Zhang Z X. A serine/threonine protein kinase encoding gene KERNEL NUMBER PER ROW6 regulates maize grain yield. Nat Commun, 2020, 11: 988.
doi: 10.1038/s41467-020-14746-7 pmid: 32080171 |
| [38] |
Zeng Z B. Precision mapping of quantitative trait loci. Genetics, 1994, 136: 1457-1468.
doi: 10.1093/genetics/136.4.1457 pmid: 8013918 |
| [39] |
Li H H, Ye G Y, Wang J K. A modified algorithm for the improvement of composite interval mapping. Genetics, 2007, 175: 361-374.
doi: 10.1534/genetics.106.066811 pmid: 17110476 |
| [1] | 艾蓉, 张春, 悦曼芳, 邹华文, 吴忠义. 玉米转录因子ZmEREB211对非生物逆境胁迫的应答[J]. 作物学报, 2023, 49(9): 2433-2445. |
| [2] | 黄钰杰, 张啸天, 陈会丽, 王宏伟, 丁双成. 玉米ZmC2s基因家族鉴定及ZmC2-15耐热功能分析[J]. 作物学报, 2023, 49(9): 2331-2343. |
| [3] | 白岩, 高婷婷, 卢实, 郑淑波, 路明. 近四十年来我国玉米大品种的历史沿革与发展趋势[J]. 作物学报, 2023, 49(8): 2064-2076. |
| [4] | 王兴荣, 张彦军, 涂奇奇, 龚佃明, 邱法展. 一个新的玉米细胞核雄性不育突变体ms6的鉴定与基因定位[J]. 作物学报, 2023, 49(8): 2077-2087. |
| [5] | 王娟, 徐相波, 张茂林, 刘铁山, 徐倩, 董瑞, 刘春晓, 关海英, 刘强, 汪黎明, 何春梅. 一个新的玉米Miniature1基因等位突变体的鉴定与遗传分析[J]. 作物学报, 2023, 49(8): 2088-2096. |
| [6] | 韦金贵, 郭瑶, 柴强, 殷文, 樊志龙, 胡发龙. 水氮减量密植玉米的产量及产量构成[J]. 作物学报, 2023, 49(7): 1919-1929. |
| [7] | 李荣, 勉有明, 侯贤清, 李培富, 王西娜. 施氮对还田秸秆腐解及养分释放、土壤肥力与玉米产量的影响[J]. 作物学报, 2023, 49(7): 2012-2022. |
| [8] | 梅秀鹏, 赵子堃, 贾欣瑶, 白洋, 李梅, 甘宇玲, 杨秋悦, 蔡一林. 热诱导转录因子ZmNF-YC13调控热胁迫应答基因提高玉米耐热性[J]. 作物学报, 2023, 49(7): 1747-1757. |
| [9] | 常丽娟, 梁晋刚, 宋君, 刘文娟, 付成平, 代晓航, 王东, 魏超, 熊梅. 转基因玉米ND207转化事件特异性定性PCR检测方法及其标准化[J]. 作物学报, 2023, 49(7): 1818-1828. |
| [10] | 王让剑, 杨军, 张力岚, 高香凤. 茶树新梢中香叶醇樱草糖苷含量的全基因组关联分析[J]. 作物学报, 2023, 49(7): 1843-1859. |
| [11] | 唐玉凤, 姚敏, 何昕, 官梅, 刘忠松, 官春云, 钱论文. 甘蓝型油菜SGR基因家族的全基因组鉴定与功能分析[J]. 作物学报, 2023, 49(7): 1829-1842. |
| [12] | 田敏, 刘新春, 潘佳佳, 梁丽静, 董雷, 刘美池, 冯宗云. 大麦籽粒纤维素、半纤维素含量全基因组关联分析[J]. 作物学报, 2023, 49(6): 1726-1732. |
| [13] | 张振博, 贾春兰, 任佰朝, 刘鹏, 赵斌, 张吉旺. 氮磷配施对夏玉米产量和叶片衰老特性的影响[J]. 作物学报, 2023, 49(6): 1616-1629. |
| [14] | 马娟, 朱卫红, 刘京宝, 宇婷, 黄璐, 郭国俊. 玉米穗长一般配合力多位点全基因组关联分析和预测[J]. 作物学报, 2023, 49(6): 1562-1572. |
| [15] | 刘佳, 邹晓悦, 马继芳, 王永芳, 董志平, 李志勇, 白辉. 谷子MAPK家族成员的鉴定及其对生物胁迫的响应分析[J]. 作物学报, 2023, 49(6): 1480-1495. |
|